8.3 Stereochemistry of Organic Compounds and Pharmaceuticals
Chirality and Stereoisomers
We turn now to the concept of chirality that formed the basis of the story about Louis Pasteur in the beginning of this chapter. Recall that the term chiral, from the Greek work for “hand,” refers to anything which cannot be superimposed on its own mirror image. Your hands, of course, are chiral—you cannot superimpose your left hand on your right, and you cannot fit your left hand into a right-handed glove (which is also a chiral object). Another way of saying this is that your hands do not have a mirror plane of symmetry: you cannot find any plane which bisects your hand in such a way that one side of the plane is a mirror image of the other side. Chiral objects do not have a plane of symmetry.
Your face, on the other hand is achiral—lacking chirality—because, some small deviations notwithstanding, you could superimpose your face onto its mirror image. If someone were to show you a mirror image photograph of your face, you could line the image up, point-for-point, with your actual face. Your face has a plane of symmetry, because the left side is the mirror image of the right side.
What Pasteur, Biot, and their contemporaries did not yet fully understand when Pasteur made his discovery of molecular chirality was the source of chirality at the molecular level. It stood to reason that a chiral molecule is one that does not contain a plane of symmetry, and thus cannot be superimposed on its mirror image. We now know that chiral molecules contain one or more chiral centres, which are almost always tetrahedral (sp3-hybridized) carbons with four different substituents. Consider the cartoon molecule A below: a tetrahedral carbon, with four different substituents denoted by balls of four different colours (for the time being, don’t worry about exactly what these substituents could be—we will see real examples very soon).
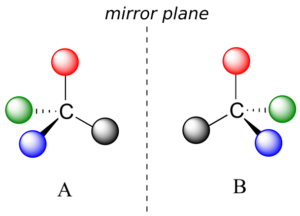
The mirror image of A, which we will call B, is drawn on the right side of the figure, and an imaginary mirror is in the middle. Notice that every point on A lines up through the mirror with the same point on B: in other words, if A looked in the mirror, it would see B looking back.
Now, if we flip compound A over and try to superimpose it point for point on compound B, we find that we cannot do it: if we superimpose any two coloured balls, then the other two are misaligned.
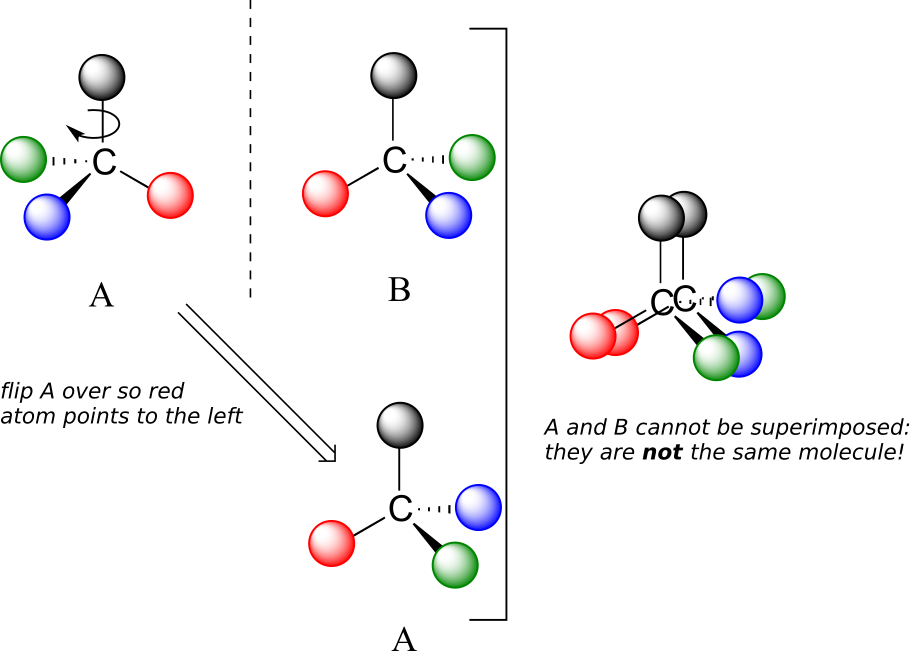
A is not superimposable on its mirror image (B), thus by definition A is a chiral molecule. It follows that B also is not superimposable on its mirror image (A), and thus it is also a chiral molecule. Also notice in the figure below (and convince yourself with models) that neither A nor B has an internal plane of symmetry.
A and B are stereoisomers: molecules with the same molecular formula and the same bonding arrangement, but a different arrangement of atoms in space. There are two types of stereoisomers: enantiomers and diastereomers. Enantiomers are pairs of stereoisomers which are mirror images of each other: thus, A and B are enantiomers. It should be self-evident that a chiral molecule will always have one (and only one) enantiomer: enantiomers come in pairs. Enantiomers have identical physical properties (melting point, boiling point, density, and so on). However, enantiomers do differ in how they interact with polarized light (we will learn more about this soon) and they may also interact in very different ways with other chiral molecules—proteins, for example.
Diastereomers are stereoisomers which are not mirror images of each other. For now, we will concentrate on understanding enantiomers, and come back to diastereomers later.
We defined a chiral centre as a tetrahedral carbon with four different substituents. If, instead, a tetrahedral carbon has two identical substituents (two black atoms in the cartoon figure below), then of course it still has a mirror image (everything has a mirror image, unless we are talking about a vampire!). However, it is superimposable on its mirror image, and has a plane of symmetry.
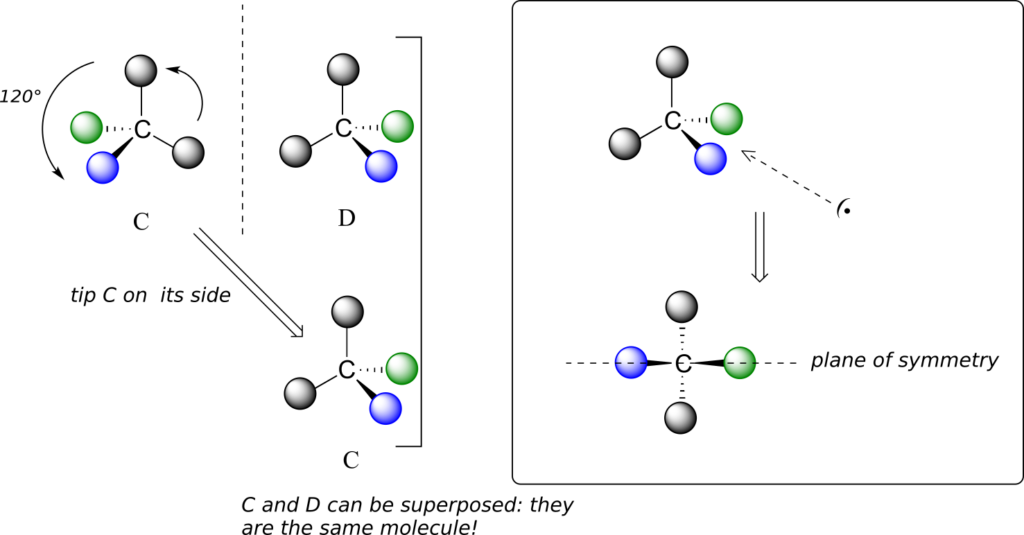
This molecule is achiral (lacking chirality). Using the same reasoning, we can see that a trigonal planar (sp2-hybridized) carbon is also not a chiral centre.
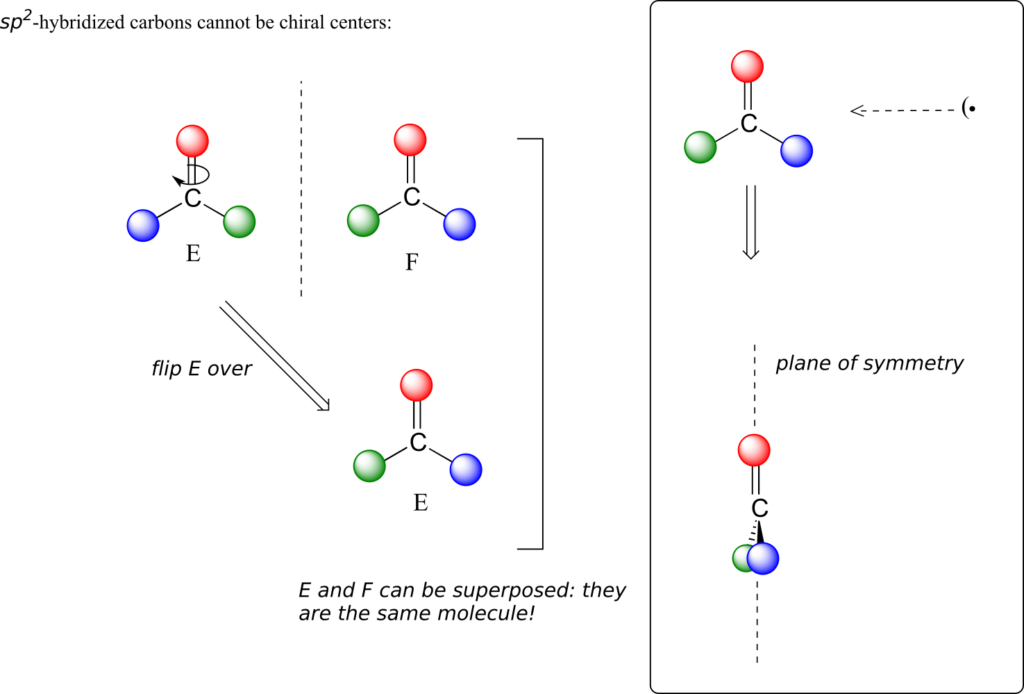
Notice that structure E can be superimposed on F, its mirror image—all you have to do is pick E up, flip it over, and it is the same as F. This molecule has a plane of symmetry, and is achiral.
Let’s apply our general discussion to real molecules. For now, we will limit our discussion to molecules with a single chiral centre. It turns out that tartaric acid, the subject of our chapter introduction, has two chiral centres, so we will come back to it later.
Consider 2-butanol, drawn in two dimensions below.
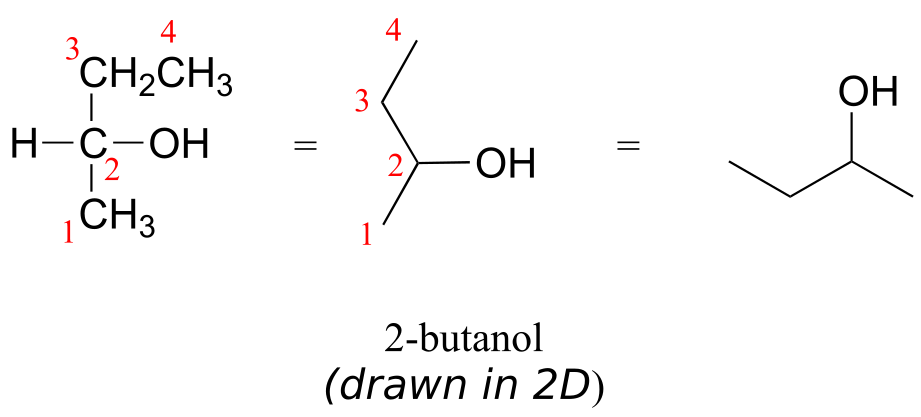
Carbon #2 is a chiral centre: it is sp3-hybridized and tetrahedral (even though it is not drawn that way above), and the four things attached to it are different: a hydrogen, a methyl (-CH3) group, an ethyl (-CH2CH3) group, and a hydroxyl (OH) group. Let’s draw the bonding at C2 in three dimensions, and call this structure A. We will also draw the mirror image of A, and call this structure B.
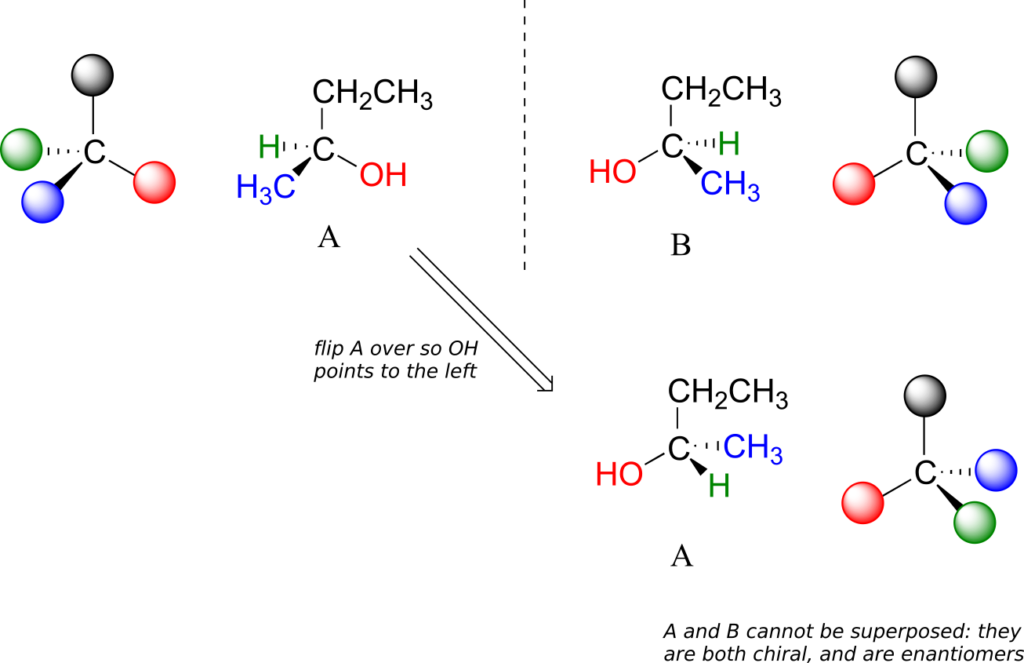
When we try to superimpose A onto B, we find that we cannot do it. A and B are both chiral molecules, and they are enantiomers of each other.
2-propanol, unlike 2-butanol, is not a chiral molecule. Carbon #2 is bonded to two identical substituents (methyl groups), and so it is not a chiral centre.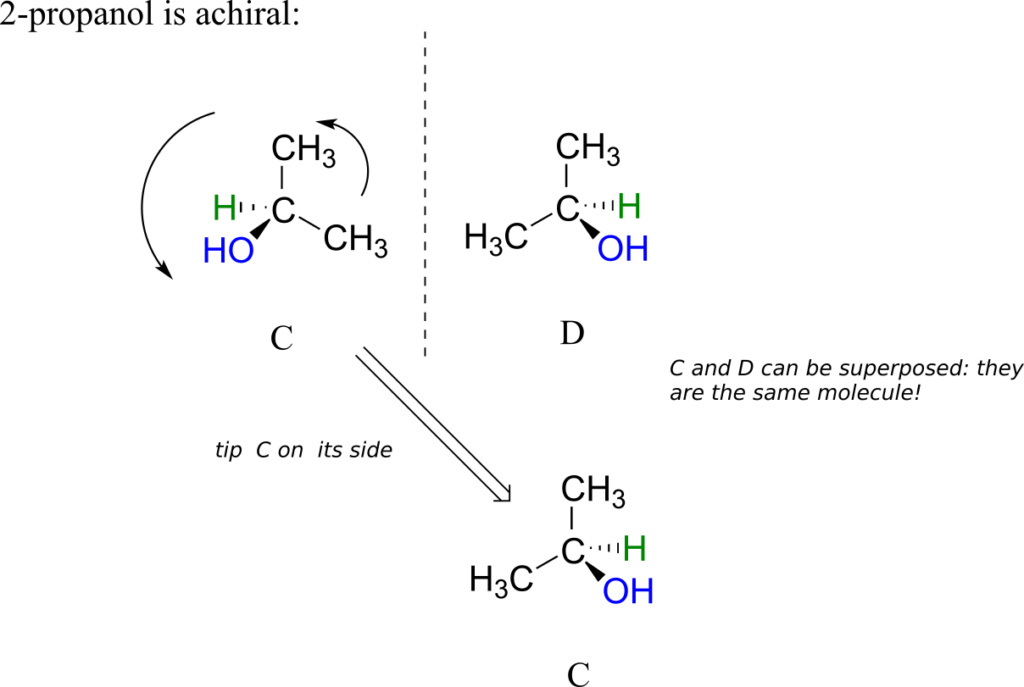
Notice that 2-propanol is superimposable on its own mirror image.
When we look at very simple molecules like 2-butanol, it is not difficult to draw out the mirror image and recognize that it is not superimposable. However, with larger, more complex molecules, this can be a daunting challenge in terms of drawing and three-dimensional visualization. The easy way to determine if a molecule is chiral is simply to look for the presence of one or more chiral centres: molecules with chiral centres will (almost always) be chiral. We insert the “almost always” caveat here because it is possible to come up with the exception to this rule—we will have more to say on this later, but don’t worry about it for now.
Here’s another trick to make your stereochemical life easier: if you want to draw the enantiomer of a chiral molecule, it is not necessary to go to the trouble of drawing the point-for-point mirror image, as we have done up to now for purposes of illustration. Instead, keep the carbon skeleton the same, and simply reverse the solid and dashed wedge bonds on the chiral carbon: that accomplishes the same thing. You should use models to convince yourself that this is true, and also to convince yourself that swapping any two substituents about the chiral carbon will result in the formation of the enantiomer.
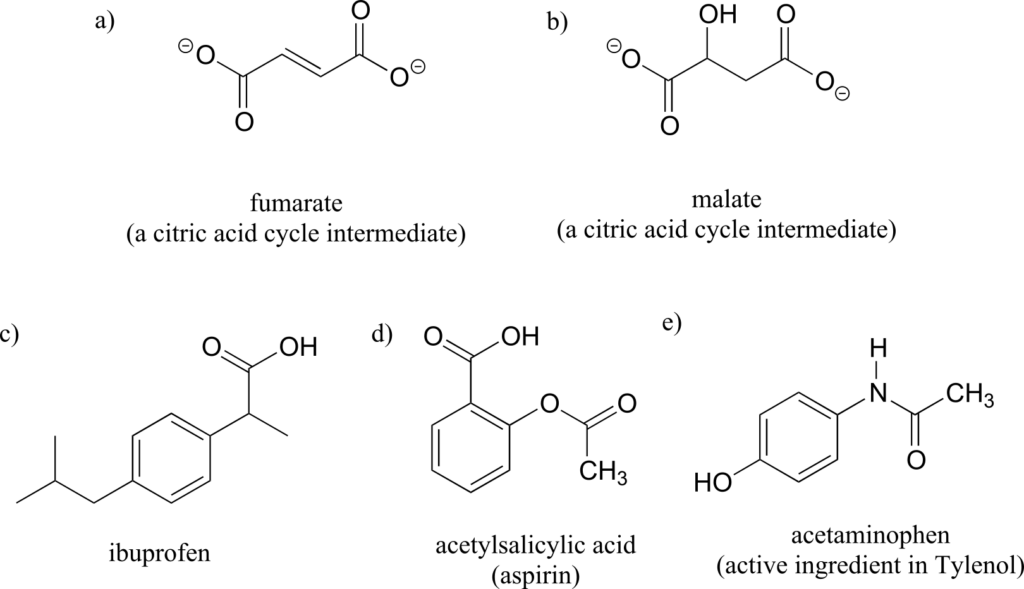
Here are four more examples of chiral biomolecules, each one shown as a pair of enantiomers, with chiral centres marked by red dots.
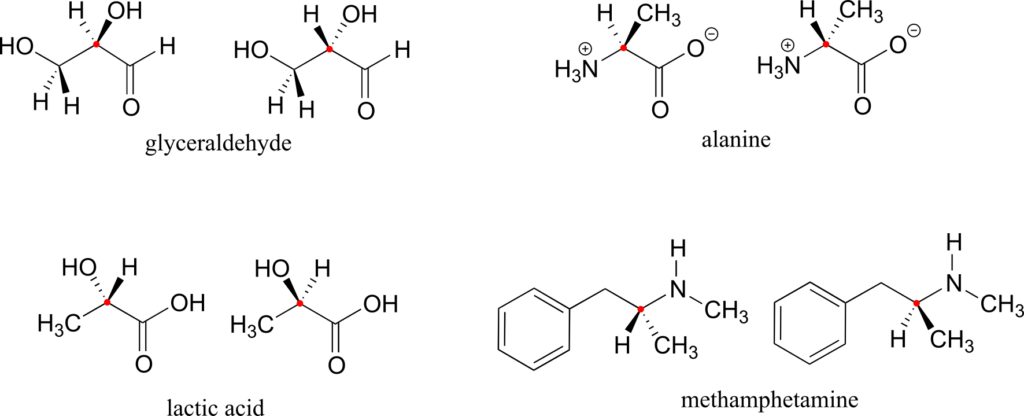
Here are some examples of achiral biomolecules—convince yourself that none of them contains a chiral centre:
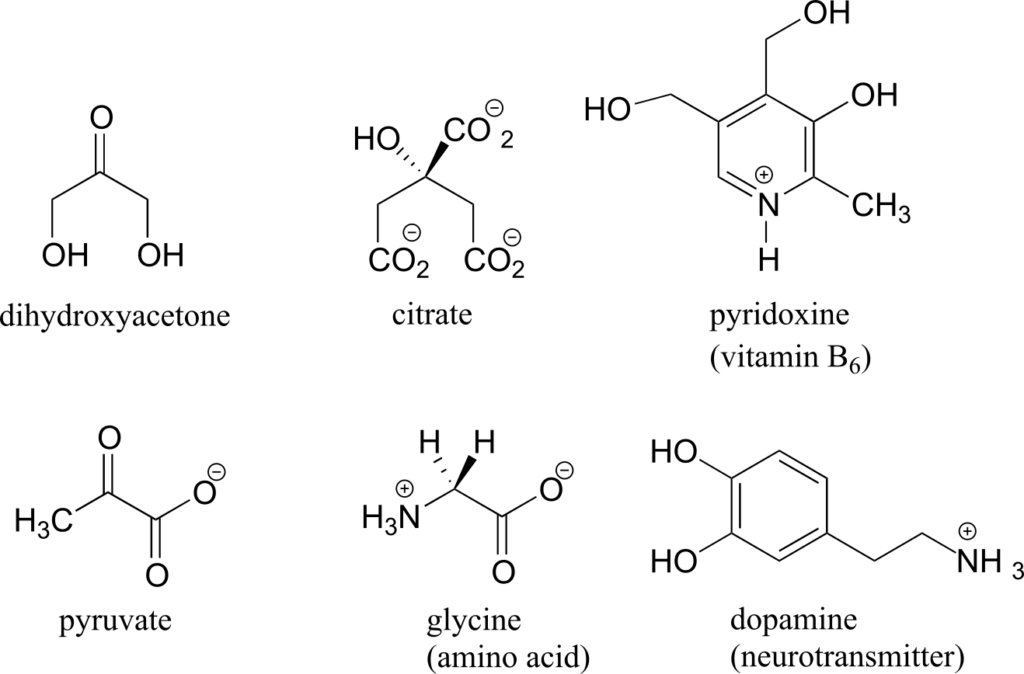
When looking for chiral centres, it is important to recognize that the question of whether or not the dashed/solid wedge drawing convention is used is irrelevant. Chiral molecules are sometimes drawn without using wedges (although obviously this means that stereochemical information is being omitted). Conversely, wedges may be used on carbons that are not chiral centres—look, for example, at the drawings of glycine and citrate in the figure above.
Can a chiral centre be something other than a tetrahedral carbon with four different substituents? The answer to this question is “yes”—however, these alternative chiral centres are very rare in the context of biological organic chemistry, and outside the scope of our discussion here.
You may also have wondered about amines: shouldn’t we consider a secondary or tertiary amine to be a chiral centre, as they are tetrahedral and attached to four different substituents, if the lone-pair electrons are counted as a “substituent”? Put another way, isn’t an amine non-superimposable on its mirror image?
The answer: yes it is, in the static picture, but in reality, the nitrogen of an amine is rapidly and reversibly inverting, or turning inside out, at room temperature.

If you have trouble picturing this, take an old tennis ball and cut it in half. Then, take one of the concave halves and flip it inside out, then back again: this is what the amine is doing. The end result is that the two “enantiomers” of the amine are actually two rapidly interconverting forms of the same molecule, and thus the amine itself is not a chiral centre. This inversion process does not take place on a tetrahedral carbon, which of course has no lone-pair electrons.
Exercise 8:
Locate all of the chiral centres (there may be more than one in a molecule). Remember, hydrogen atoms bonded to carbon usually are not drawn in the line structure convention—but they are still there!
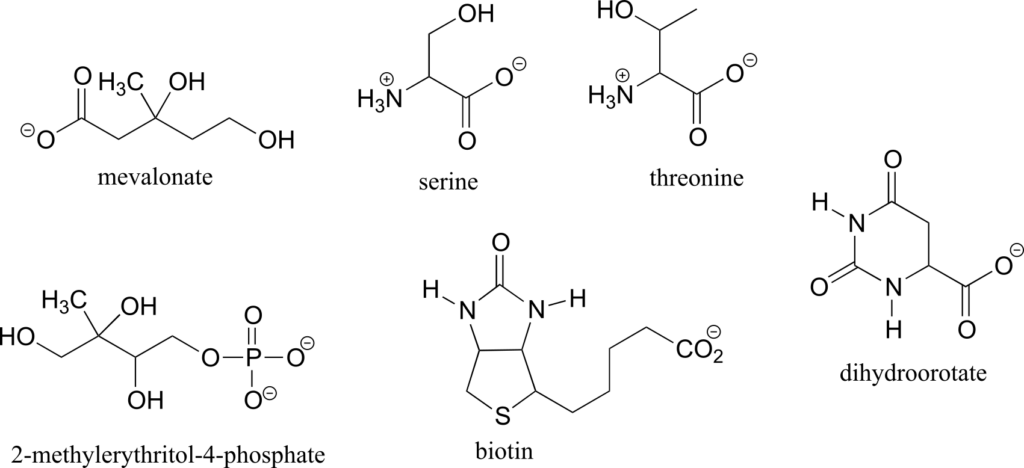
Exercise 9:
- Draw two enantiomers of i) mevalonate and ii) serine.
- Are the two 2-butanol structures below enantiomers?
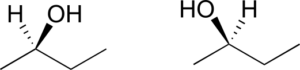
Exercise 10:
Label the molecules below as chiral or achiral, and locate all chiral centres.
Video tutorials: Chirality (Khan Academy)
Video tutorials: Enantiomers (Khan Academy)
Labelling Chiral Centres
Chemists need a convenient way to distinguish one stereoisomer from another. The Cahn-Ingold-Prelog system is a set of rules that allows us to unambiguously define the stereochemical configuration of any stereocenter, using the designations “R” (from the Latin rectus, meaning right-handed) or “S” (from the Latin sinister, meaning left-handed).
The rules for this system of stereochemical nomenclature are, on the surface, fairly simple.
Rules for assigning an R/S designation to a chiral centre:
- Assign priorities to the four substituents, with #1 being the highest priority and #4 the lowest. Priorities are based on the atomic number.
- Trace a circle from #1 to #2 to #3.
- Determine the orientation of the #4 priority group. If it is oriented into the plane of the page (away from you), go to step 4a. If it is oriented out of the plane of the page (toward you), go to step 4b.
- (#4 group pointing away from you): a clockwise circle in part 2 corresponds to the R configuration, while a counterclockwise circle corresponds to the S configuration.
- (#4 group pointing toward you): a clockwise circle in part 2 corresponds to the S configuration, while a counterclockwise circle corresponds to the R configuration.
We’ll use the 3-carbon sugar glyceraldehyde as our first example. The first thing that we must do is to assign a priority to each of the four substituents bound to the chiral centre. We first look at the atoms that are directly bonded to the chiral centre: these are H, O (in the hydroxyl), C (in the aldehyde), and C (in the CH2OH group).
Assigning R/S configuration to glyceraldehyde:
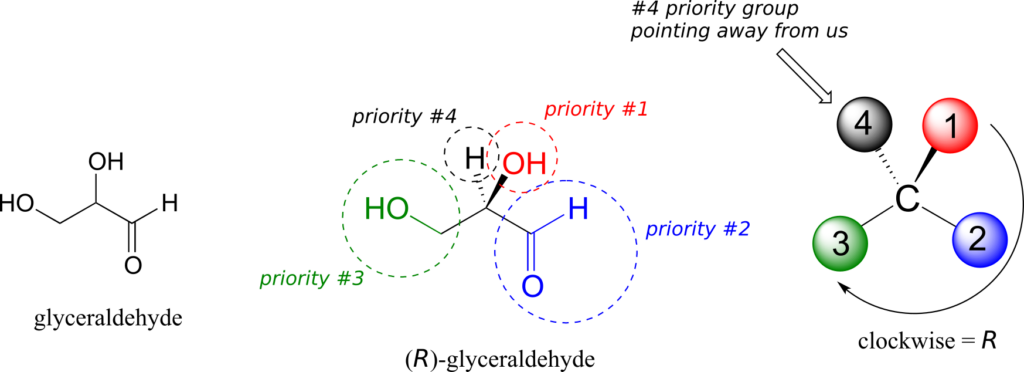
Two priorities are easy: hydrogen, with an atomic number of 1, is the lowest (#4) priority, and the hydroxyl oxygen, with atomic number 8, is priority #1. Carbon has an atomic number of 6. Which of the two “C” groups is priority #2, the aldehyde or the CH2OH? To determine this, we move one more bond away from the chiral centre: for the aldehyde we have a double bond to an oxygen, while on the CH2OH group we have a single bond to an oxygen. If the atom is the same, double bonds have a higher priority than single bonds. Therefore, the aldehyde group is assigned #2 priority and the CH2OH group the #3 priority.
With our priorities assigned, we look next at the #4 priority group (the hydrogen) and see that it is pointed back away from us, into the plane of the page—thus step 4a from the procedure above applies. Then, we trace a circle defined by the #1, #2, and #3 priority groups, in increasing order. The circle is clockwise, which by step 4a tells us that this carbon has the “R” configuration, and that this molecule is (R)-glyceraldehyde. Its enantiomer, by definition, must be (S)-glyceraldehyde.
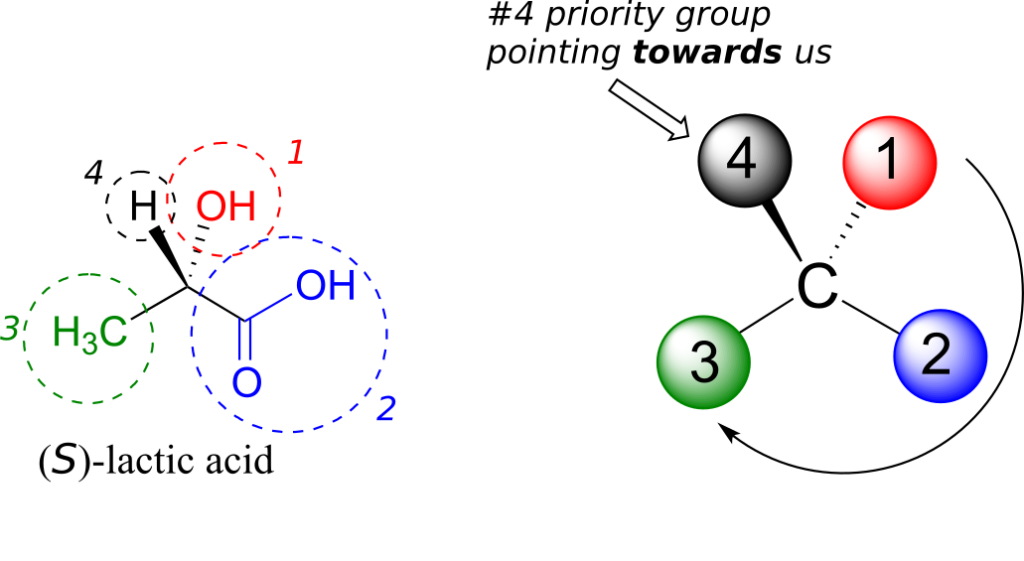
Next, let’s look at one of the enantiomers of lactic acid and determine the configuration of the chiral centre. Clearly, H is the #4 substituent and OH is #1. Owing to its three bonds to oxygen, the carbon on the acid group takes priority #2, and the methyl group takes #3. The #4 group, hydrogen, happens to be drawn pointing toward us (out of the plane of the page) in this figure, so we use step 4b: The circle traced from #1 to #2 to #3 is clockwise, which means that the chiral centre has the S configuration.
Interactive model: “Chiral Molecules R or S” (ChemTube3D)
Video tutorial: Cahn-Ingold-Prelog System for Naming Enantiomers (Khan Academy)
The drug thalidomide is an interesting—but tragic—case study in the importance of stereochemistry in drug design. First manufactured by a German drug company and prescribed widely in Europe and Australia in the late 1950s as a sedative and remedy for morning sickness in pregnant women, thalidomide was soon implicated as the cause of devastating birth defects in babies born to women who had taken it. Thalidomide contains a chiral centre, and thus exists in two enantiomeric forms. It was marketed as a racemic mixture: in other words, a 50:50 mixture of both enantiomers.
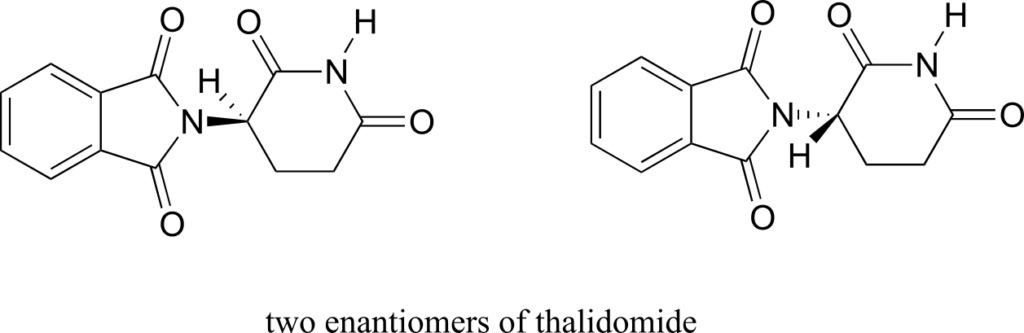
Let’s try to determine the stereochemical configuration of the enantiomer on the left. Of the four bonds to the chiral centre, the #4 priority is hydrogen. The nitrogen group is #1, the carbonyl side of the ring is #2, and the –CH2 side of the ring is #3.
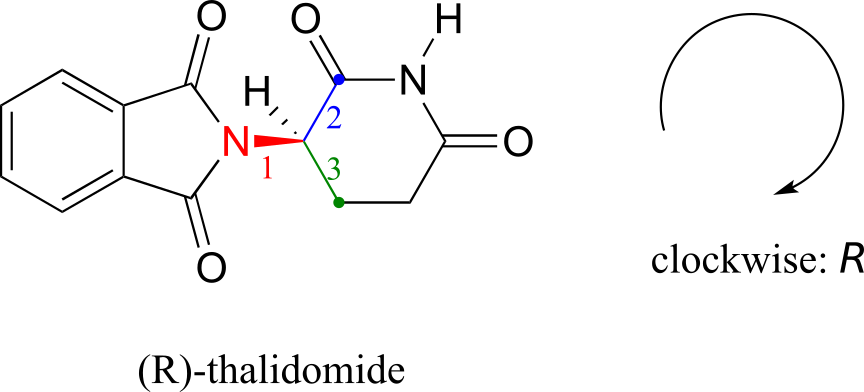
The hydrogen is shown pointing away from us, and the prioritized substituents trace a clockwise circle: this is the R enantiomer of thalidomide. The other enantiomer, of course, must have the S configuration.
Interactive model of (S)-thalidomide (JSmol)
Although scientists are still unsure today how thalidomide works, experimental evidence suggests that it was actually the R enantiomer that had the desired medical effects, while the S enantiomer caused the birth defects. Even with this knowledge, however, pure (R)-thalidomide is not safe, because enzymes in the body rapidly convert between the two enantiomers.
As a historical note, thalidomide was never approved for use in the United States. This was thanks in large part to the efforts of Dr. Frances Kelsey, a Food and Drug officer who, at peril to her career, blocked its approval due to her concerns about the lack of adequate safety studies, particularly with regard to the drug’s ability to enter the bloodstream of a developing fetus. Unfortunately, though, at that time clinical trials for new drugs involved widespread and unregulated distribution to doctors and their patients across the country, so families in the U.S. were not spared from the damage caused.
Very recently, a close derivative of thalidomide has become legal to prescribe again in the United States, with strict safety measures enforced, for the treatment of a form of blood cancer called multiple myeloma. In Brazil, thalidomide is used in the treatment of leprosy—but despite safety measures, children are still being born with thalidomide-related defects.
Exercise 11:
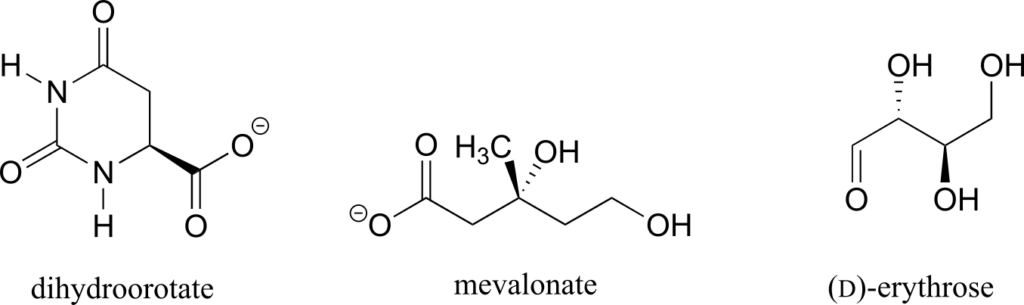
Determine the stereochemical configurations of the chiral centres in the biomolecules shown below.
Exercise 12:
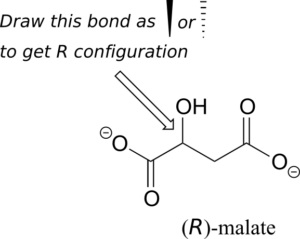
Should the (R) enantiomer of malate have a solid or dashed wedge for the C-O bond in the figure below?
Exercise 13:
Using solid or dashed wedges to show stereochemistry, draw the (R) enantiomer of ibuprofen and the (S) enantiomer of 2-methylerythritol-4-phosphate.
Optical Activity
Chiral molecules have an interesting optical property. You may know from studying physics that light waves are oscillating electric and magnetic fields. In ordinary light, the oscillation is randomly oriented in an infinite number of planes. When ordinary light is passed through a polarizer, all planes of oscillation are filtered out except one, resulting in plane-polarized light.
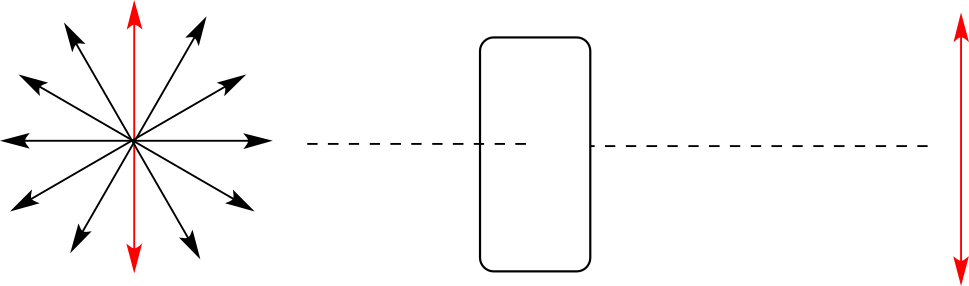
A beam of plane-polarized light, when passed through a sample of a chiral compound, interacts with the compound in such a way that the angle of oscillation will rotate. This property is called optical activity.
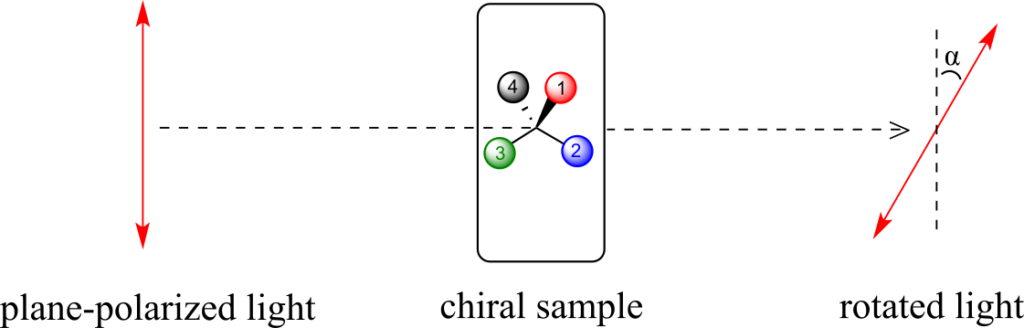
If a compound rotates plane polarized light in the clockwise (+) direction, it is said to be dextrorotatory, while if it rotates light in the counterclockwise (-) direction it is levorotatory. (We mentioned L- and D-amino acids in the previous section: the L-amino acids are levorotatory.) The magnitude of the observed optical activity is dependent on temperature, the wavelength of light used, solvent, concentration of the chiral sample, and the path length of the sample tube (path length is the length that the plane-polarized light travels through the chiral sample). Typically, optical activity measurements are made in a 1-decimetre (10 cm) path-length sample tube at 25 °C, using as a light source the so-called “D-line” from a sodium lamp, which has a wavelength of 589 nm. The specific rotation [a] of a pure chiral compound at 25° is expressed by the expression:
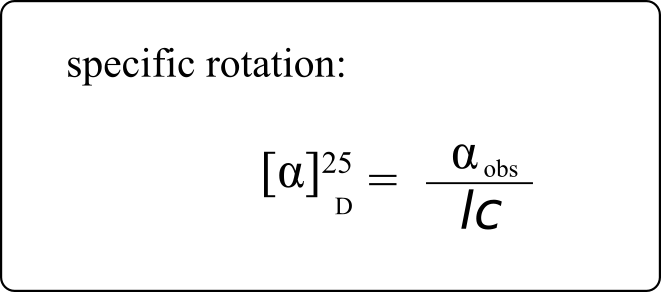
. . . where αobs is the observed rotation, l is path length in decimetres, and c is the concentration of the sample in grams per 100 mL. In other words, the specific rotation of a chiral compound is the optical rotation that is observed when 1 g of the compound is dissolved in enough of a given solvent to make 100 mL solution, and the rotation is measured in a 1-dm cuvette at 25 °C using light from a sodium lamp.
Every chiral molecule has a characteristic specific rotation, which is recorded in the chemical literature as a physical property just like melting point or density. Different enantiomers of a compound will always rotate plane-polarized light with an equal but opposite magnitude. (S)-ibuprofen, for example, has a specific rotation of +54.5° (dextrorotatory) in methanol, while (R)-ibuprofen has a specific rotation of -54.5°. There is no relationship between chiral compound’s R/S designation and the direction of its specific rotation. For example, the S enantiomer of ibuprofen is dextrorotatory, but the S enantiomer of glyceraldehyde is levorotatory.
A 50:50 mixture of two enantiomers (a racemic mixture) will have no observable optical activity, because the two optical activities cancel each other out. In a structural drawing, a “squiggly” bond from a chiral centre indicates a mixture of both R and S configurations.

Chiral molecules are often labelled according to whether they are dextrorotatory or levorotatory as well as by their R/S designation. For example, the pure enantiomers of ibuprofen are labelled (S)-(+)-ibuprofen and (R)-(-)-ibuprofen, while (±)-ibuprofen refers to the racemic mixture, which is the form in which the drug is sold to consumers.
Video tutorial: Optical Activity (Khan Academy)
Exercise 14:
The specific rotation of (R)-limonene is +11.5° in ethanol. What is the expected observed rotation of a sample of 6.00-g (S)-limonene dissolved in ethanol to a total volume of 80.0 mL in a 1.00-dm (10.0 cm) pathlength cuvette?
Exercise 15:
The specific rotation of (S)-carvone is +61°, measured “neat” (pure liquid sample, no solvent). The optical rotation of a mixture of R and S carvone is measured at -23°. Which enantiomer is in excess in the mixture?
All of the 20 natural amino acids except glycine have a chiral centre at their α-carbon. Virtually all of the amino acids found in nature, both in the form of free amino acids or incorporated into peptides and proteins, have what is referred to in the biochemical literature as the “L” configuration:
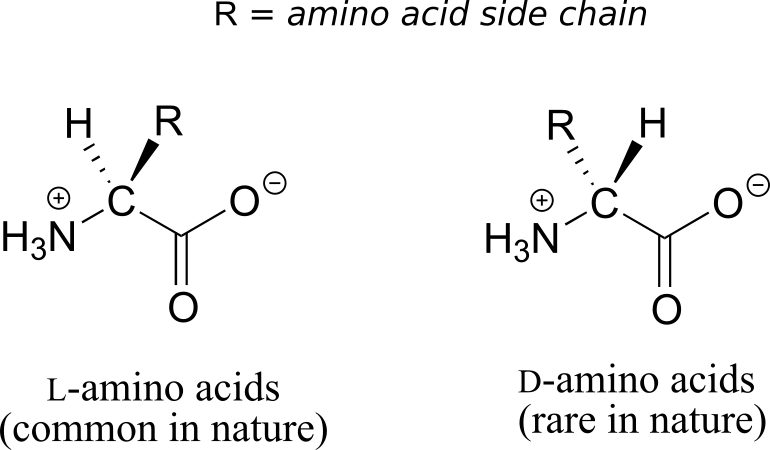
The “L” indicates that these amino acid stereoisomers are levorotatory. All but one of the 19 L-amino acids have S stereochemistry at the α-carbon, using the rules of the R/S naming system.
Interactive model: “Chiral Molecules R or S” (ChemTube3D)
D-amino acids (the D stands for dextrorotatory) are very rare in nature.
Exercise 16:
Which L-amino acid has the R configuration? Refer to the structures of all 20 common amino acids.
Compounds with Multiple Chiral Centres
So far, we have been analyzing compounds with a single chiral centre. Next, we turn our attention to those which have multiple chiral centres. We’ll start with some stereoisomeric four-carbon sugars with two chiral centres.
To avoid confusion, we will simply refer to the different stereoisomers by capital letters.
Look first at compound A, below. Both chiral centres have the R configuration (you should confirm this for yourself!). The mirror image of compound A is compound B, which has the S configuration at both chiral centres. If we were to pick up compound A, flip it over, and put it next to compound B, we would see that they are not superimposable (again, confirm this for yourself with your models!). A and B are nonsuperimposable mirror images: in other words, enantiomers.
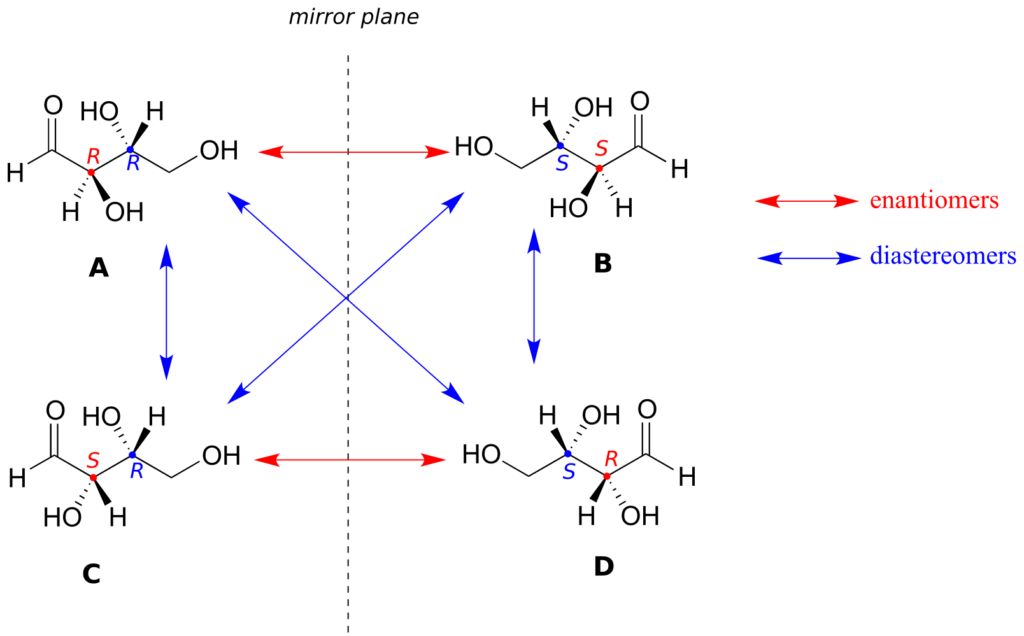
Now, look at compound C, in which the configuration is S at chiral centre 1 and R at chiral centre 2. Compounds A and C are stereoisomers: they have the same molecular formula and the same bond connectivity, but a different arrangement of atoms in space (recall that this is the definition of the term “stereoisomer”). However, they are not mirror images of each other (confirm this with your models!), and so they are not enantiomers. By definition, they are diastereomers of each other.
Notice that compounds C and B also have a diastereomeric relationship, by the same definition.
So, compounds A and B are a pair of enantiomers, and compound C is a diastereomer of both of them. Does compound C have its own enantiomer? Compound D is the mirror image of compound C, and the two are not superimposable. Therefore, C and D are a pair of enantiomers. Compound D is also a diastereomer of compounds A and B.
This can also seem very confusing at first, but there some simple shortcuts to analyzing stereoisomers:
Stereoisomer Shortcuts
If all of the chiral centres are of opposite R/S configuration between two stereoisomers, they are enantiomers.
If at least one, but not all of the chiral centres are opposite between two stereoisomers, they are diastereomers.
(Note: these shortcuts do not take into account the possibility of additional stereoisomers due to alkene groups; we will come to that later.)
Here’s another way of looking at the four stereoisomers, where one chiral centre is associated with red and the other blue. Pairs of enantiomers are stacked together.
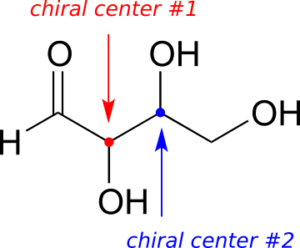
The four possible configurations:

We know, using the shortcut above, that the enantiomer of RR must be SS—both chiral centres are different. We also know that RS and SR are diastereomers of RR, because in each case one—but not both—chiral centres are different.
Now, let’s extend our analysis to a sugar molecule with three chiral centres. Going through all the possible combinations, we come up with eight total stereoisomers—four pairs of enantiomers.
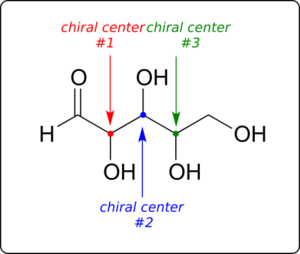

Let’s draw the RRR stereoisomer. Being careful to draw the wedge bonds correctly so that they match the RRR configurations, we get:
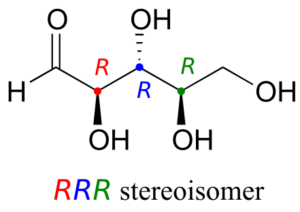
Now, using the above drawing as our model, drawing any other stereoisomer is easy. If we want to draw the enantiomer of RRR, we don’t need to try to visualize the mirror image, we just start with the RRR structure and invert the configuration at every chiral centre to get SSS.
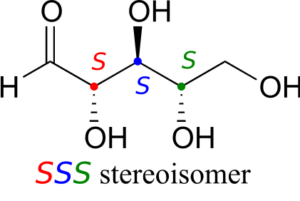
Try making models of RRR and SSS and confirm that they are in fact nonsuperimposable mirror images of each other.
There are six diastereomers of RRR. To draw one of them, we just invert the configuration of at least one, but not all three, of the chiral centres. Let’s invert the configuration at chiral centre 1 and 2, but leave chiral centre 3 unchanged. This gives us the SSR configuration.
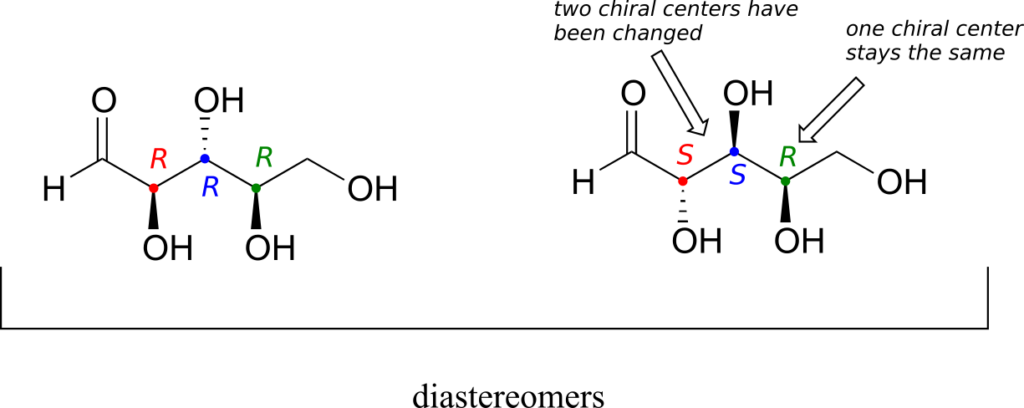
One more definition at this point: diastereomers which differ at only a single chiral centre are called epimers. For example, RRR and SRR are epimers:
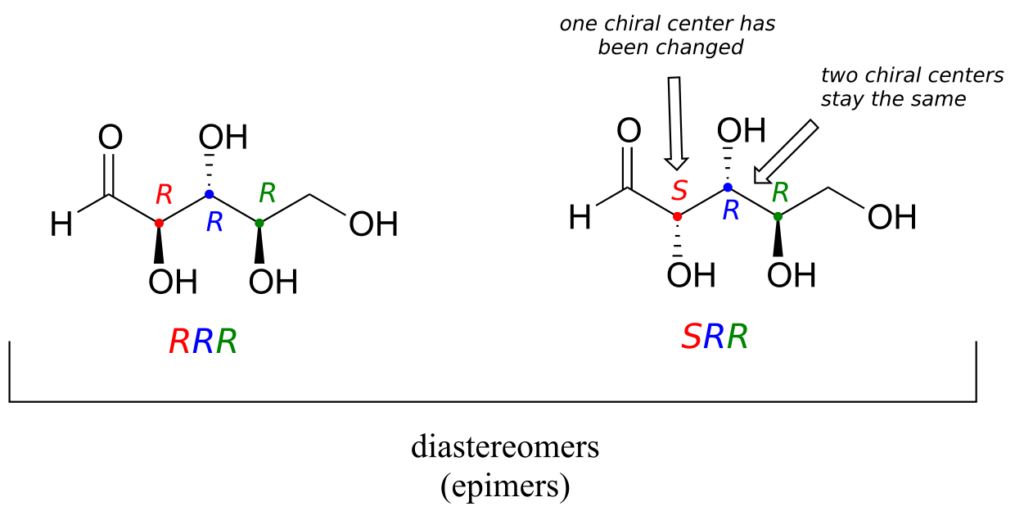
The RRR and SSR stereoisomers shown earlier are diastereomers but not epimers because they differ at two of the three chiral centres.
Exercise 17:
- Draw the structure of the enantiomer of the SRS stereoisomer of the sugar used in the previous example.
- List (using the XXX format, not drawing the structures) all of the epimers of SRS.
- List all of the stereoisomers that are diastereomers, but not epimers, of SRS.
The epimer term is useful because in biochemical pathways, compounds with multiple chiral centres are isomerized at one specific centre by enzymes known as epimerases. Two examples of epimerase-catalyzed reactions are below.
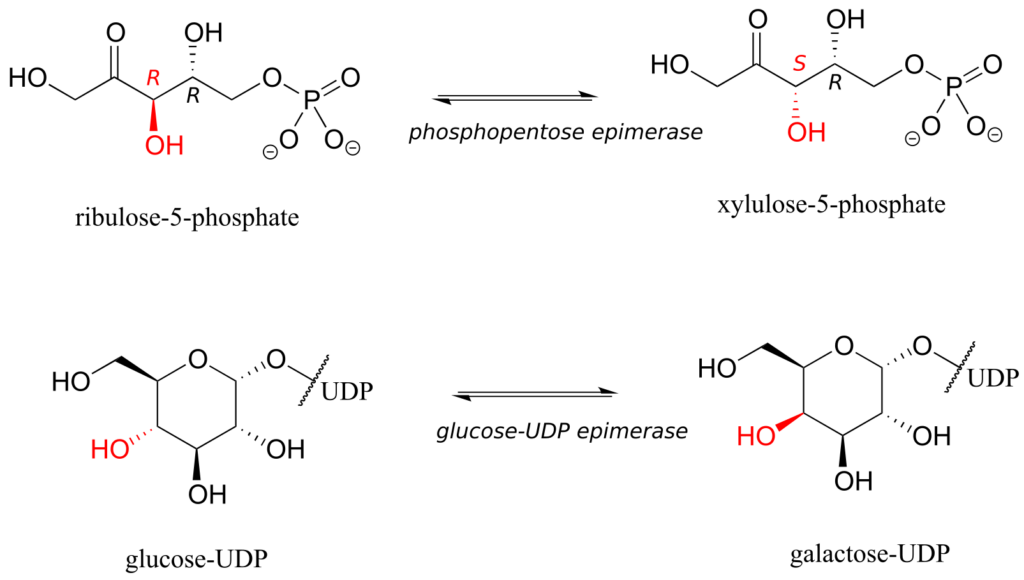
We know that enantiomers have identical physical properties and equal but opposite magnitude specific rotation. Diastereomers, in theory at least, have different physical properties—we stipulate “in theory” because sometimes the physical properties of two or more diastereomers are so similar that it is very difficult to distinguish between them. In addition, the specific rotation values of diastereomers are unrelated—they could be the same sign or opposite signs, similar in magnitude or very dissimilar.
Exercise 18:
The sugar below is one of the stereoisomers that we have been discussing.
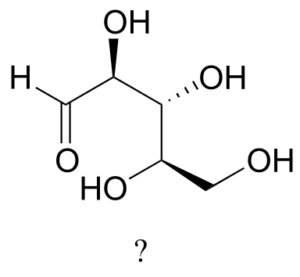
The only problem is, it is drawn with the carbon backbone in a different orientation from what we have seen. Determine the configuration at each chiral centre to determine which stereoisomer it is.
Exercise 19:
Draw the enantiomer of the xylulose-5-phosphate structure in the previous figure.
Exercise 20:
The structure of the amino acid D-threonine, drawn without stereochemistry, is shown below. D-threonine has the (S) configuration at both of its chiral centres. Draw D-threonine, its enantiomer, and its two diastereomers.
Here is some more practice in identifying isomeric relationships. D-glucose is the monosaccharide that serves as the entrance point for the glycolysis pathway and as a building block for the carbohydrate biopolymers starch and cellulose. The “D” in D-glucose stands for dextrarotatory and is part of the specialized nomenclature system for sugars, which we will not concern ourselves with here. The open-chain structure of the sugar is shown below.
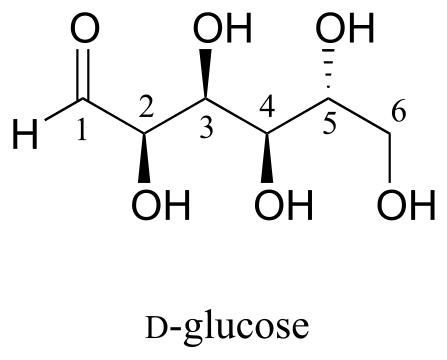
Because D-glucose has four chiral centres, it can exist in a total of 24 = 16 different stereoisomeric forms: it has one enantiomer and 14 diastereomers.
Now, let’s compare the structures of the two sugars D-glucose and D-gulose, and try to determine their relationship.

The two structures have the same molecular formula and the same connectivity, therefore they must be stereoisomers. They each have four chiral centres, and the configuration is different at two of these centres (at carbons #3 and #4). They are diastereomers.
Now, look at the structures of D-glucose and D-mannose.

Here, everything is the same except for the configuration of the chiral centre at carbon #2. The two sugars differ at only one of the four chiral centres, so again they are diastereomers, and more specifically they are epimers.
D-glucose and L-glucose are enantiomers, because they differ at all four chiral centres.
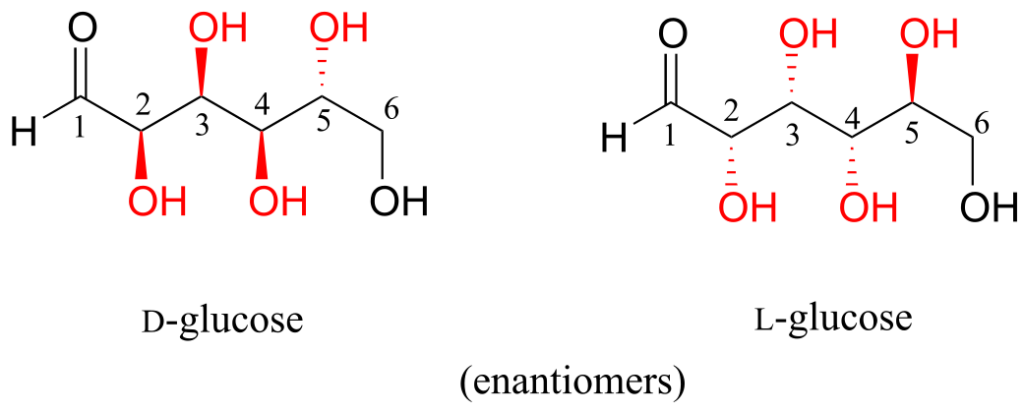
D-glucose is the enantiomer commonly found in nature.
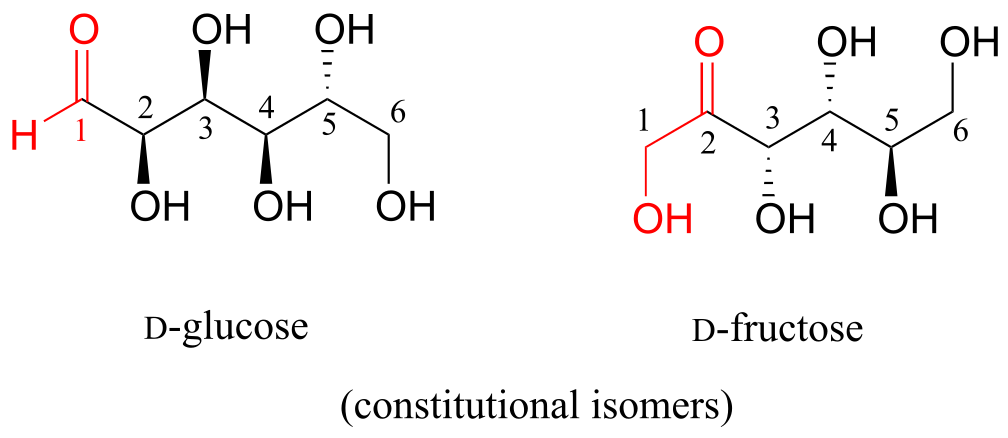
D-glucose and D-fructose are not stereoisomers, because they have different bonding connectivity: glucose has an aldehyde group, while fructose has a ketone. The two sugars do, however, have the same molecular formula, so by definition they are constitutional isomers.
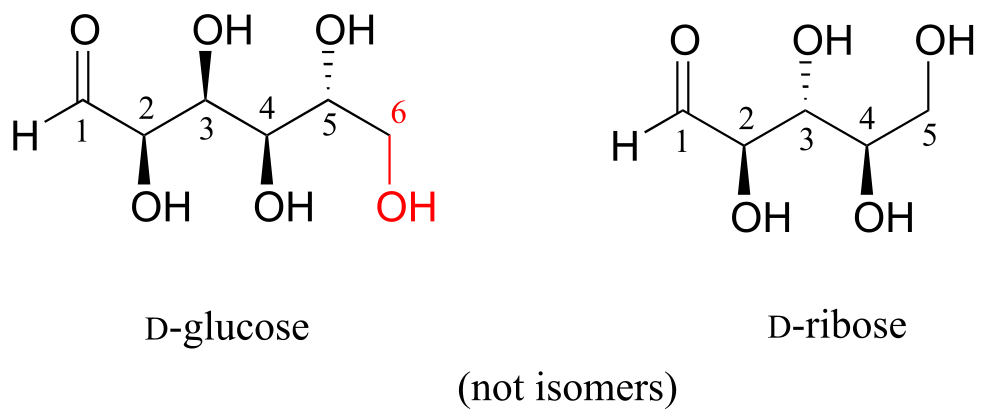
D-glucose and D-ribose are not isomers of any kind, because they have different molecular formulas.
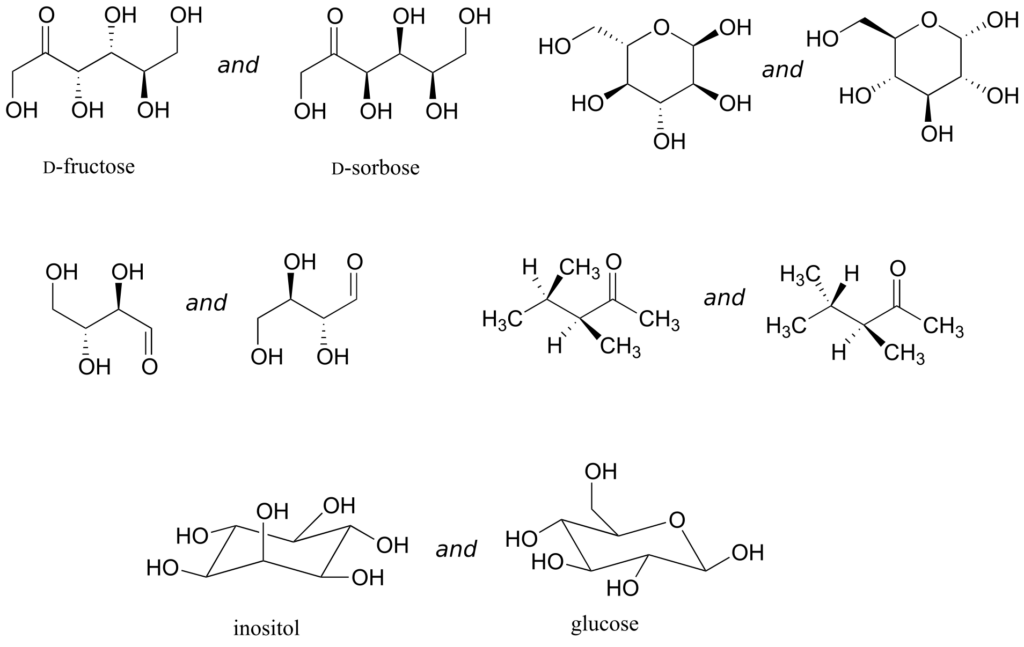
Exercise 21:
Identify the relationship between each pair of structures. Your choices are: not isomers, constitutional isomers, diastereomers but not epimers, epimers, enantiomers, or same molecule.
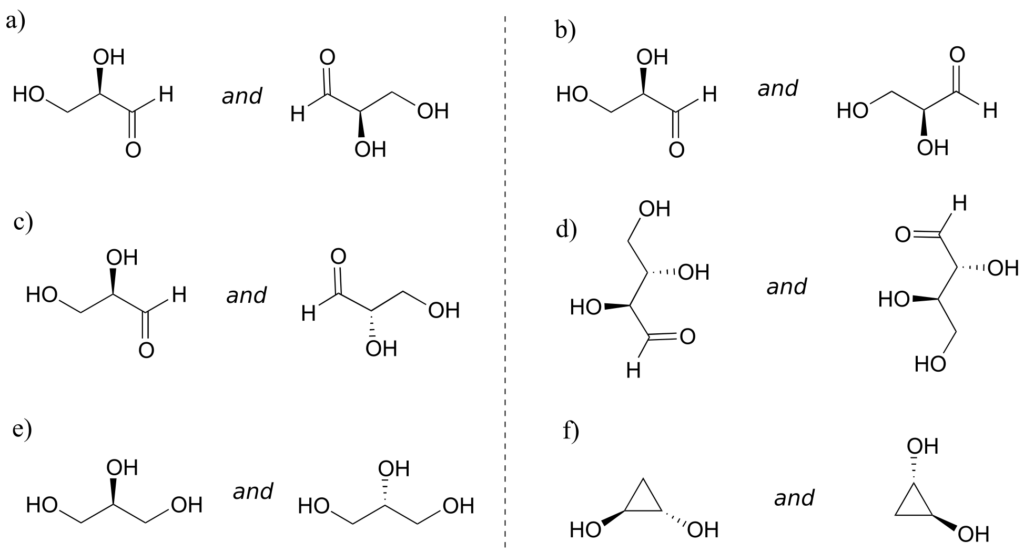
Exercise 22:
Identify the relationship between each pair of structures. Hint: figure out the configuration of each chiral centre.
Meso Compounds
The levorotatory and dextrorotatory forms of tartaric acid studied by Louis Pasteur were, as we now know, the (S,S) and (R,R) enantiomers, respectively:
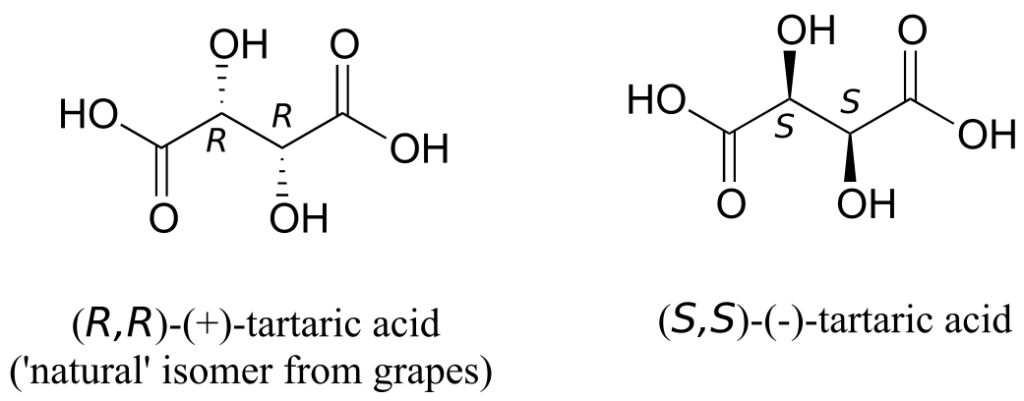
What the 19th-century chemists referred to as “acide racemique“ was just that: a racemic mixture of the R,R and S,S enantiomers, the racemization a result of how the natural R,R isomer had been processed.
But tartaric acid has two chiral centres: shouldn’t there be another pair of enantiomers?
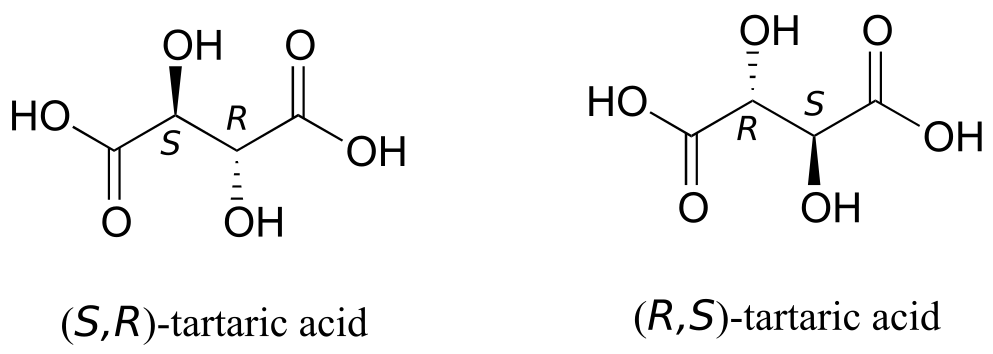
Interactive model: “Tartaric Acid – Newman Projections” (ChemTube3D)
There in fact is another stereoisomer of tartaric acid—but only one. The two structures above are actually superimposable on one another: they are the exact same molecule. The figure below illustrates this, and also that the structure has a plane of symmetry. However, you should be sure to build models and confirm these assertions for yourself.
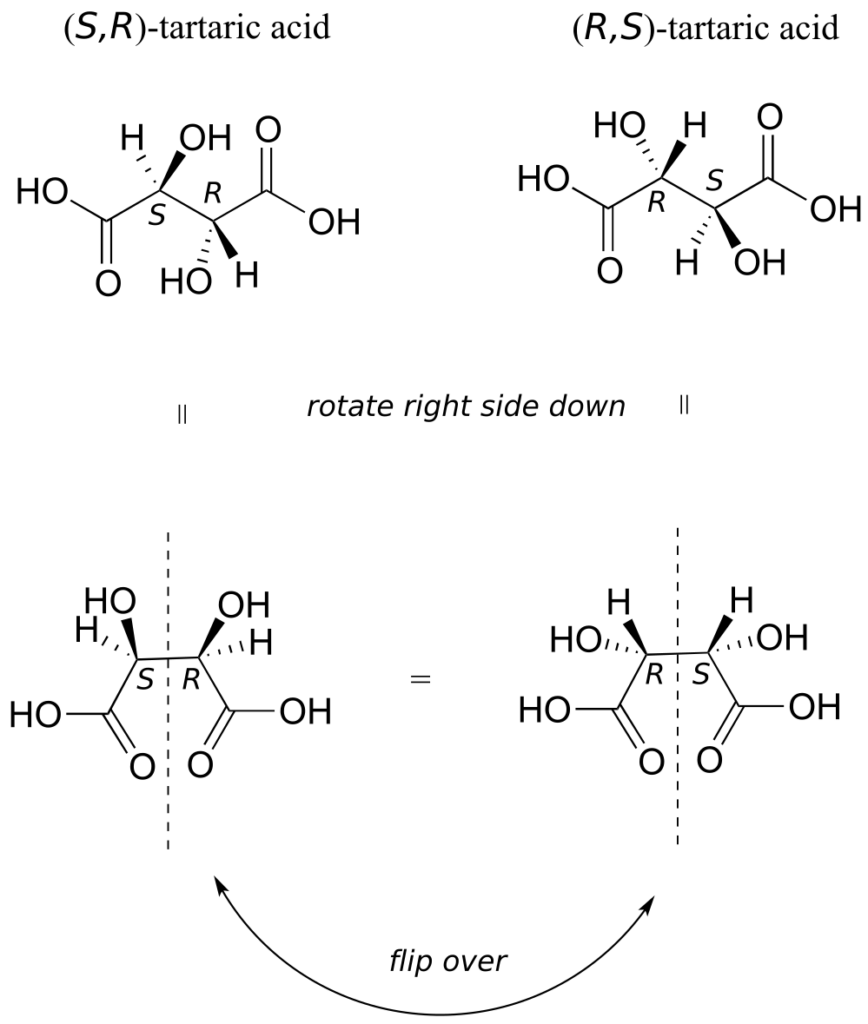
This tartaric acid isomer is an achiral diastereomer of both the levorotatory and the dextrorotatory isomers. It is a special case, called a meso compound: it has two apparent chiral centres but due to its internal symmetry it is not in fact chiral, and does not exhibit optical activity. Note that the meso form of tartaric acid did not play a part in Pasteur’s experiments.
There are many more possible examples of meso compounds, but they really can be considered “exceptions to the rule” and quite rare in biologically relevant chemistry.
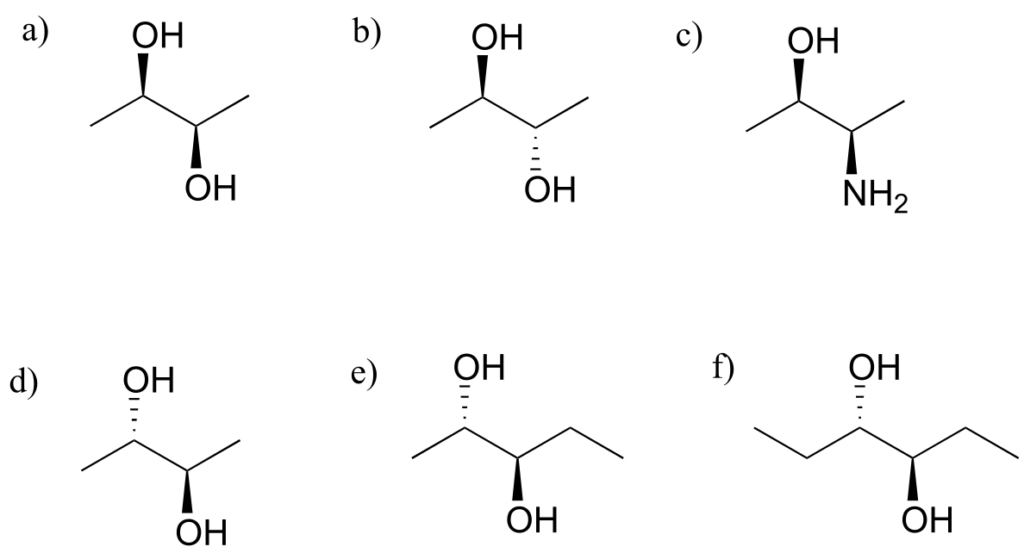
Exercise 23:
Which of the following compounds are meso? Hint: build models, and then try to find a conformation in which you can see a plane of symmetry.
Video tutorial: Stereoisomers, Enantiomers, Diastereomers, Constitutional Isomers and Meso Compounds (Khan Academy)
Fischer and Haworth Projections
When reading the chemical and biochemical literature, you are likely to encounter several different conventions for drawing molecules in three dimensions, depending on the context of the discussion. While organic chemists prefer to use the dashed/solid wedge convention to show stereochemistry, biochemists often use drawings called Fischer projections and Haworth projections to discuss and compare the structure of sugar molecules.
Fisher projections show sugars in their open-chain form. In a Fischer projection, the carbon atoms of a sugar molecule are connected vertically by solid lines, while carbon-oxygen and carbon-hydrogen bonds are shown horizontally. Stereochemical information is conveyed by a simple rule: vertical bonds point into the plane of the page, while horizontal bonds point out of the page.
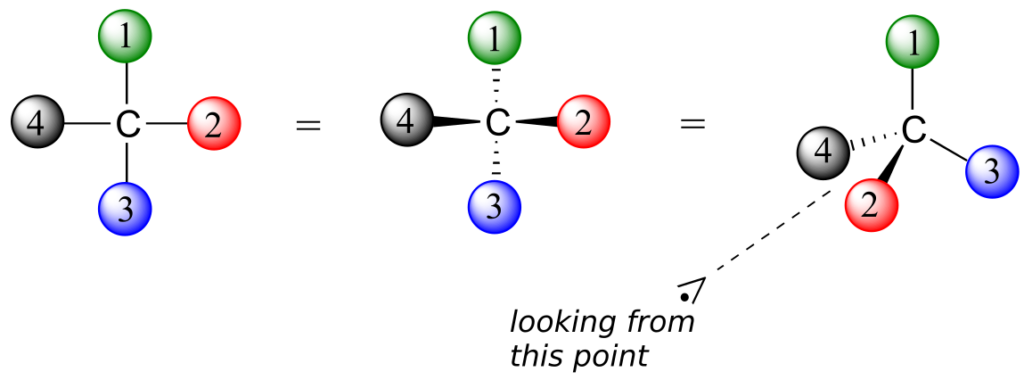
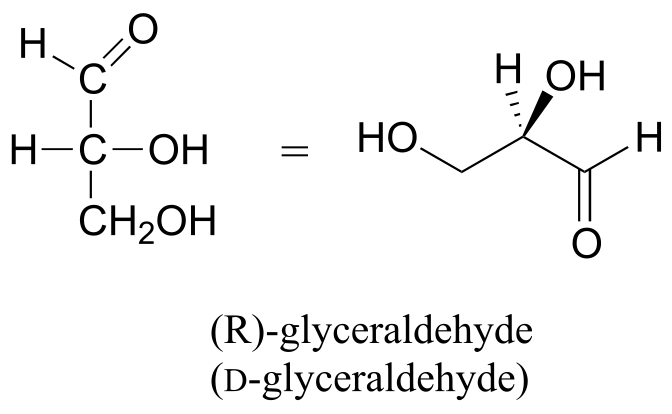
Below are two different representations of (R)-glyceraldehyde, the smallest sugar molecule (also called D-glyceraldehyde in the stereochemical nomenclature used for sugars):
Below are three representations of the open-chain form of D-glucose: in the conventional Fischer projection (A), in the “line structure” variation of the Fischer projection in which carbons and hydrogens are not shown (B), and finally in the “zigzag” style (C) that is preferred by organic chemists.
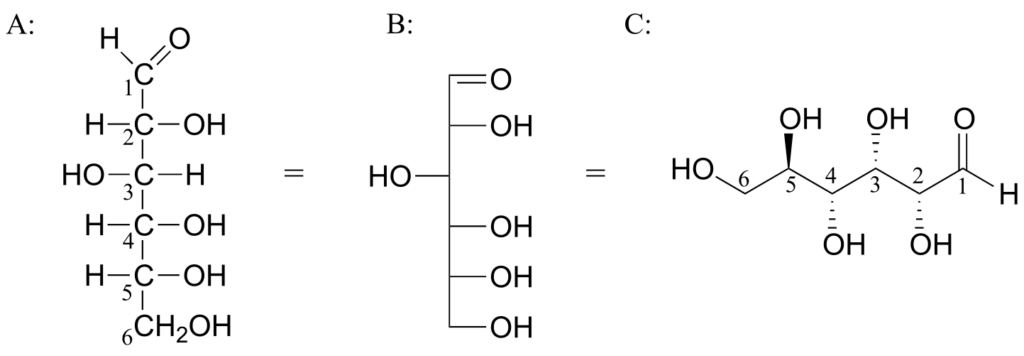
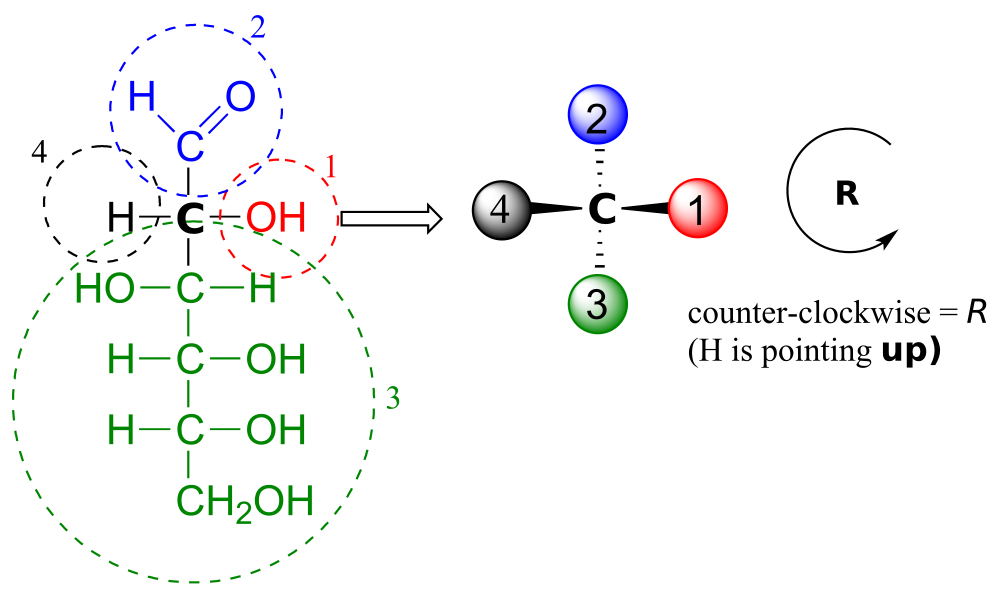
Care must be taken when “translating” Fischer projection structures into “zigzag” format—it is easy to get the stereochemistry wrong. Probably the best way to make a translation is to simply assign R/S configurations to each stereocentre, and proceed from there. When deciding whether a stereocentre in a Fischer projection is R or S, realize that the hydrogen, in a horizontal bond, is pointing towards you—therefore, a counterclockwise circle means R, and a clockwise circle means S (the opposite of when the hydrogen is pointing away from you).
Fischer projections are useful when looking at many different diastereomeric sugar structures, because the eye can quickly pick out stereochemical differences according to whether a hydroxyl group is on the left or right side of the structure.
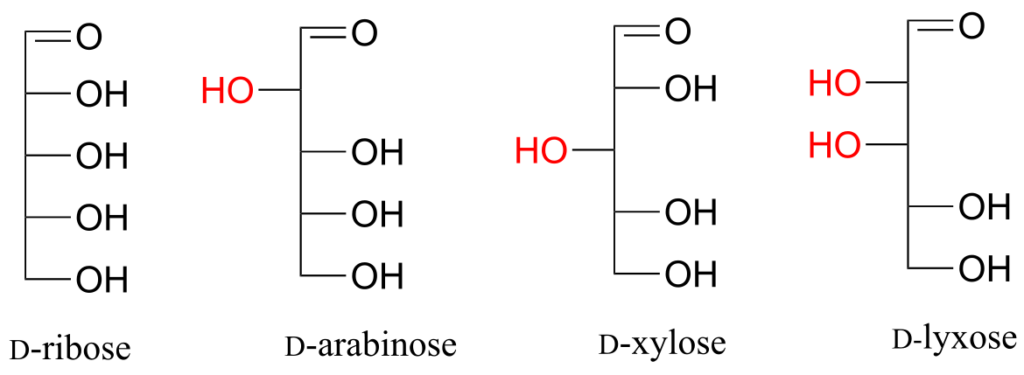
Exercise 24:
Draw “zigzag” structures (using the solid/dash wedge convention to show stereochemistry) for the four sugars in the figure above. Label all stereocentres R or S. To make it easy to check your answers, draw your structures using the framework below.
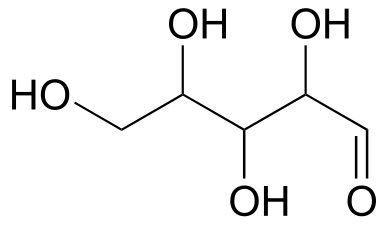
While Fischer projections are used for sugars in their open-chain form, Haworth projections are often used to depict sugars in their cyclic forms. The b diastereomer of the cyclic form of glucose is shown below in three different depictions, with the Haworth projection in the middle.
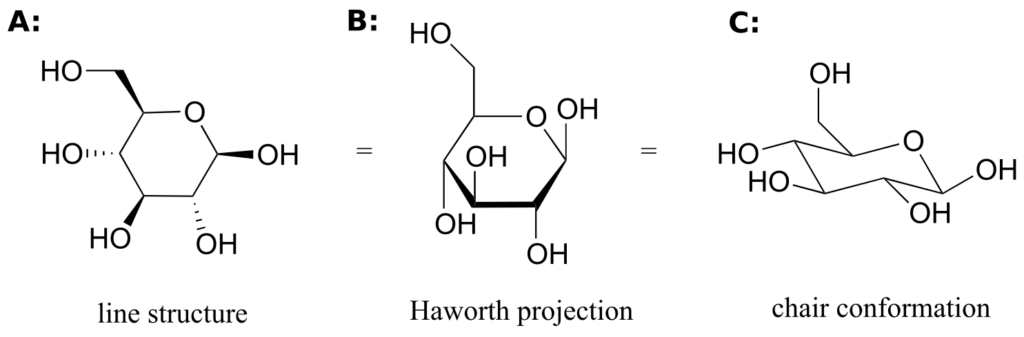
Notice that although a Haworth projection is a convenient way to show stereochemistry, it does not provide a realistic depiction of conformation. To show both conformation and stereochemistry, you must draw the ring in the chair form, as in structure C above.
Stereochemistry of Alkenes
When we talk about stereochemistry, we are not always talking about chiral compounds and chiral centres. Consider cis– and trans-2-butene:
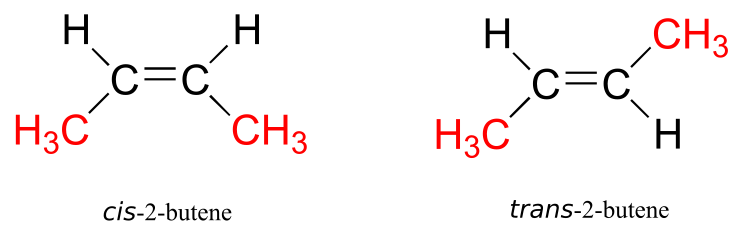
Each can be superimposed on its own mirror image, and neither is chiral (also, note the lack of a chiral centre!). However, they both have the same molecular formula and the same bonding connectivity, so by definition they are stereoisomers of each other. Because they are not mirror images, they must be diastereomers. An alkene group which can exist in two stereoisomeric forms is referred to as stereogenic.
Alkene groups in natural unsaturated fatty acids are normally cis, but trans-fatty acids (which are thought to be associated with heart disease and other health problems) are found in some food products.
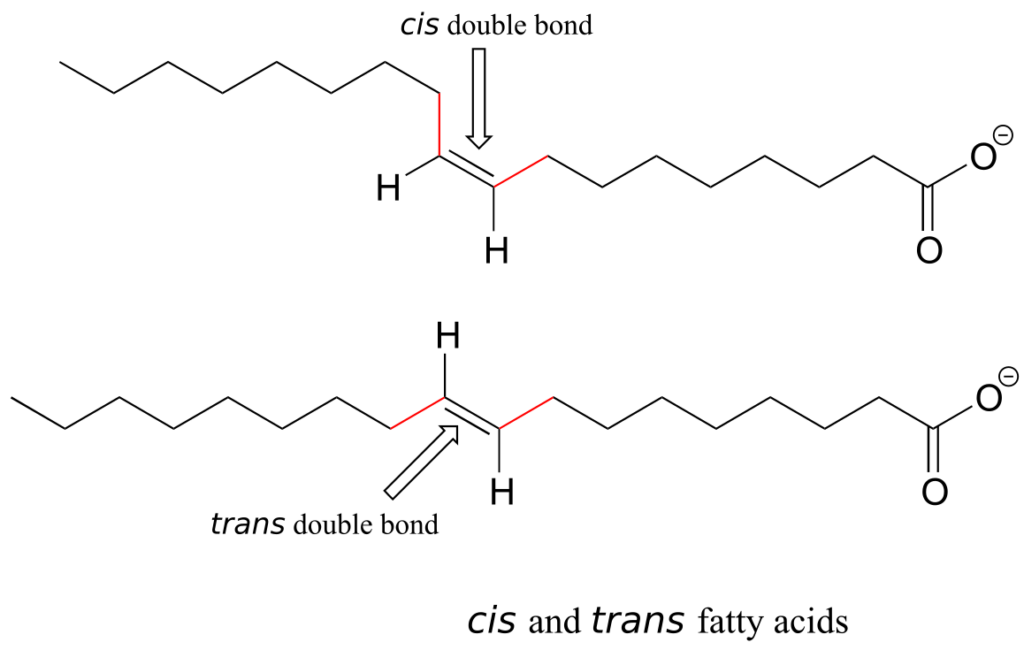
Retinal is a light-sensitive molecule, derived from vitamin A, that is found in the rod cells of the eye. When light enters the eye through the retina, one form of retinal is converted to a diastereomer when a cis double bond is converted to trans. This changes the shape of the molecule and the way that it binds to the vision protein rhodopsin, which in turn initiates a chain of events that leads to a signal being sent to the vision centre of the brain.

While the terms cis and trans are quite clear in the examples above, in some cases they can be ambiguous, and a more rigorous stereochemical designation is required. To unambiguously designate alkene stereochemistry, it is best to use the designators “E” and “Z” rather than trans and cis. To use this naming system, we first decide which is the higher-priority group on each carbon of the double bond, using the same priority rules that we learned for the R/S system. If the higher-priority groups are on the same side of the double bond, it is a Z-alkene, and if they are on the opposite side it is an E-alkene. A memory device that many students find helpful is the phrase “Z = zame zide.”
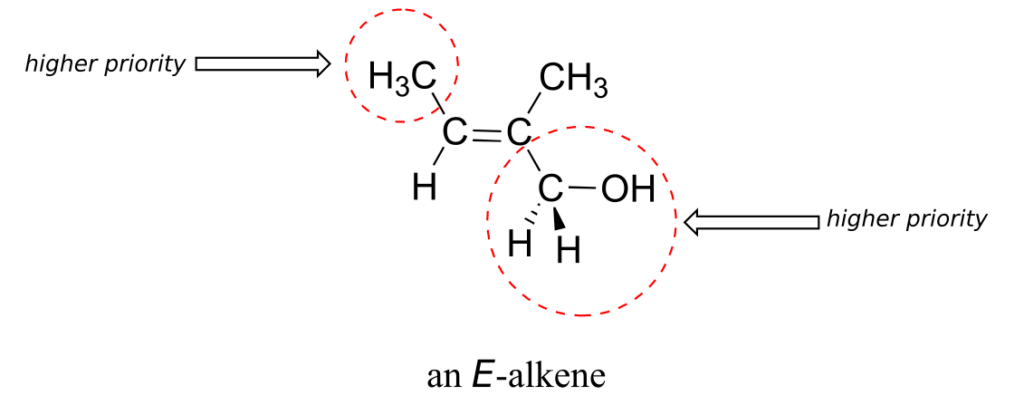
Shown below is an example of an E-alkene: notice that, although the two methyl groups are on the same side relative to one another, the alkene has E stereochemistry according to the rules of the E/Z system because one of the methyl groups takes a higher priority (relative to a hydrogen) and the other takes lower priority (relative to a primary alcohol). The cis/trans terms would be ambiguous for this compound.
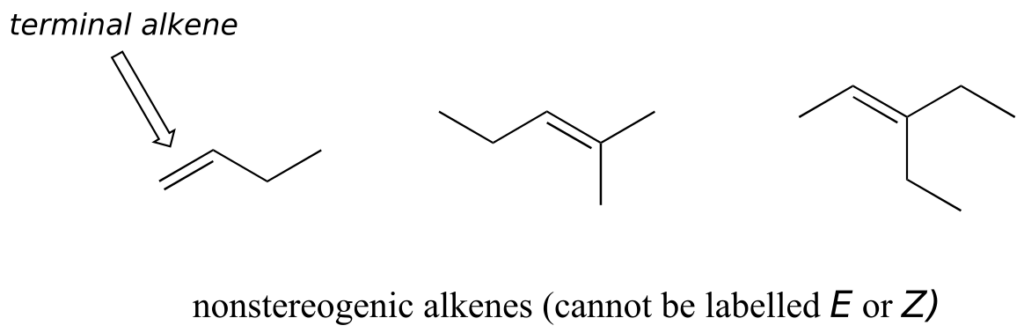
Not all alkenes can be labelled E or Z: if one (or both) of the double-bonded carbons has identical substituents, the alkene is not stereogenic, and thus cannot be assigned an E or Z configuration. Terminal alkenes, in which one of the alkene carbons is bonded to two hydrogen atoms, are the most commonly seen type of nonstereogenic alkene.
Video tutorial: E/Z System (Khan Academy)
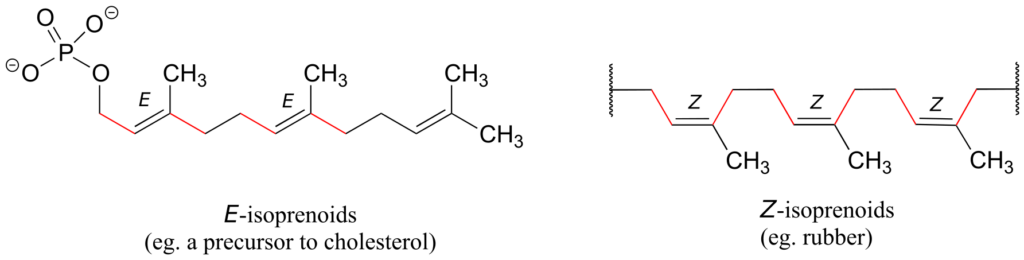
Natural rubber is a polymer composed of five-carbon isoprenoid building blocks linked with Z stereochemistry. The same isoprenoid building blocks can also be connected with E stereochemistry, leading to a polymer that is a precursor to cholesterol and many other natural isoprenoid compounds found in all forms of life.
Alkenes located inside a five- or six-membered ring, such as in cyclohexene, are not generally labelled E or Z, simply because the closed geometry of the ring allows for only one stereochemical possibility. (E)-cyclohexene is not physically possible, so it is not necessary to include the (Z) designator for cyclohexene. Larger rings, however, can hypothetically have E or Z alkene groups: two actual examples are included in Exercise 25 below.
As a general rule, alkenes with the bulkiest groups on opposite sides of the double bond are more stable, due to reduced steric strain. The trans (E) diastereomer of 2-butene, for example, is slightly lower in energy than the cis (Z) diastereomer, as seen by their relative heats of hydrogenation to butane.
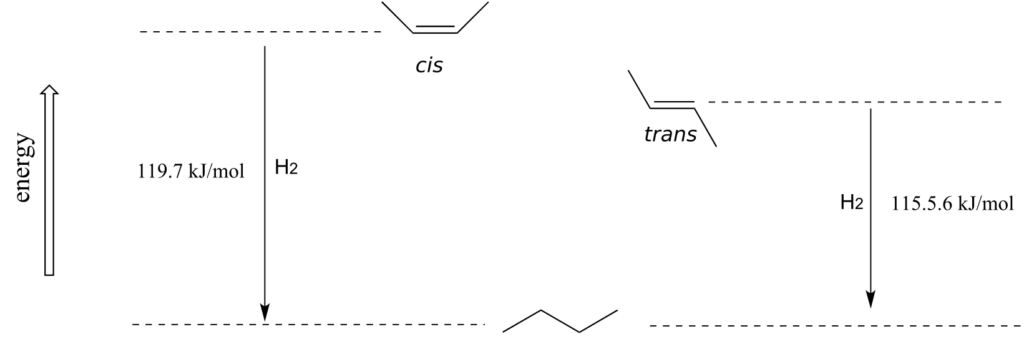
Exercise 25:
Label the alkene groups below as E, Z, or N (for a nonstereogenic alkene).
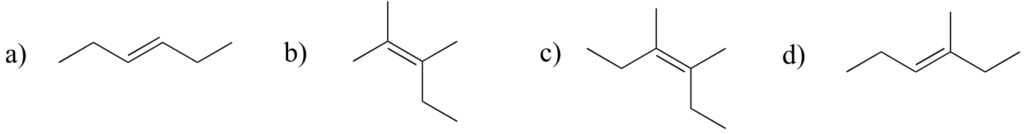
Exercise 26:
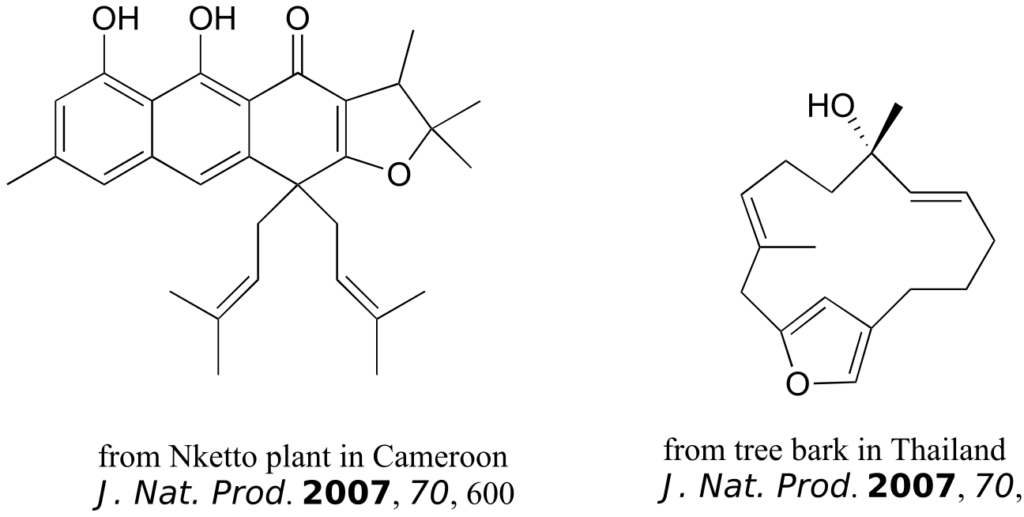
The compounds shown below were all isolated from natural sources and their structures reported in a 2007 issue of the Journal of Natural Products, an American Chemical Society publication. Label all alkene groups that are not inside 5- or 6-membered rings as E, Z, or N (for a nonstereogenic alkene).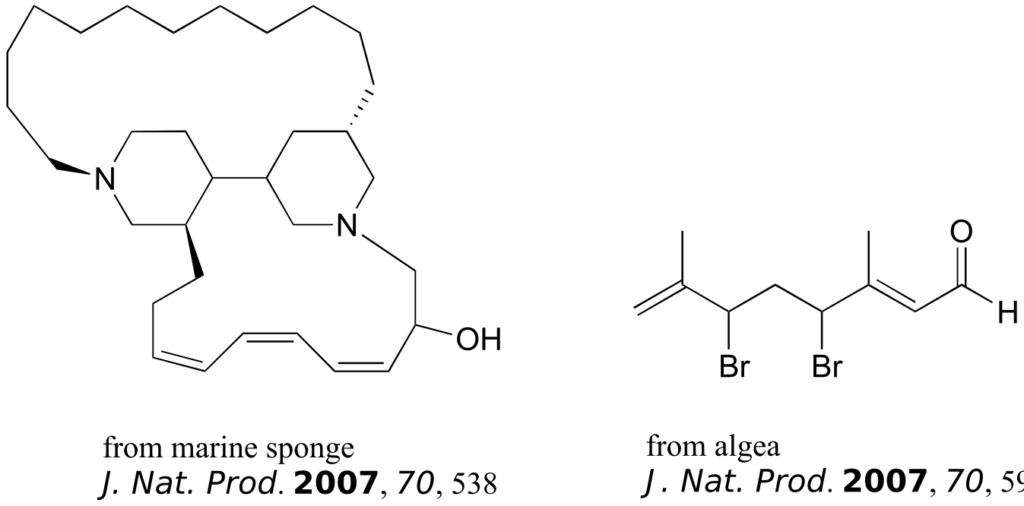
How do we know how many stereoisomers are possible for a given structure? There is actually a straightforward way to figure this out. All we need to do is count the number of chiral centres and stereogenic alkene groups, then use this following rule:
Number of stereoisomeric forms = 2n
. . . where n = the number of chiral centres plus the number of stereogenic alkene groups.
(The rare exception to this rule is when a meso form is possible—in this case, the rule becomes 2n-1.)
Consider, for example, a molecule with two chiral centres and one stereogenic alkene. By the rule stated above, we know right away that there must be eight possible stereoisomers. Drawing out all the possibilities, we see:
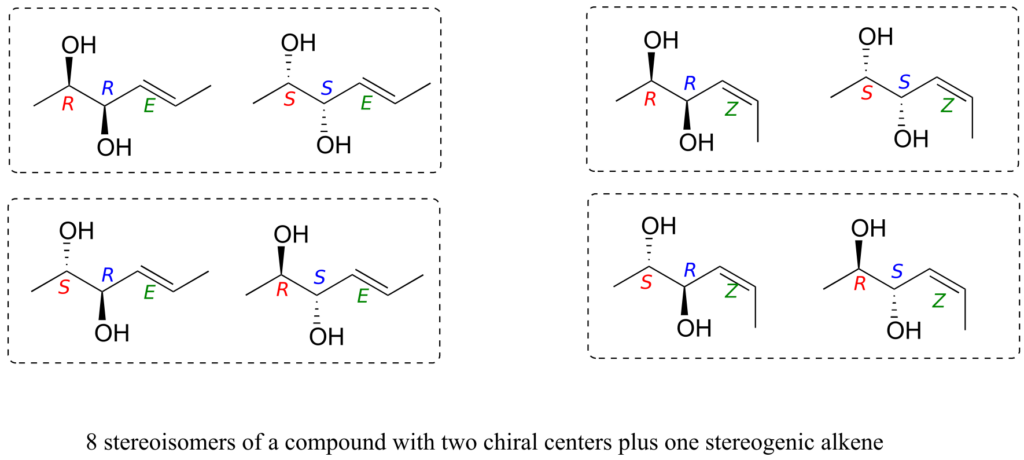
We see that, for example, RRE has one enantiomer, the SSE compound. The six other stereoisomers are all diastereomers of RRE.
It needs to be stressed that the enantiomer of the RRE compound is the SSE compound, not the SSZ compound. Remember, the E/Z relationship is diastereomeric, not enantiomeric. Use models to convince yourself that the RRE and the SSE isomers are mirror images of each other, while RRE and SSZ compounds are not. In general, to get the enantiomer of a compound, we invert all chiral centres but leave all stereogenic alkenes the same.
Exercise 27:
Draw the enantiomer of each of the compounds below, and assign configurations to all chiral centres and stereogenic alkenes. How many diastereomers are possible for each of the structures you drew?
Stereochemistry in Biology and Medicine
While challenging to understand and visualize, the stereochemistry concepts we have explored in this chapter are integral to the study of living things. The vast majority of biological molecules contain chiral centres and/or stereogenic alkene groups. Most importantly, proteins are chiral, which of course includes all of the enzymes which catalyze the chemical reactions of a cell, the receptors which transmit information within or between cells, and the antibodies which bind specifically to potentially harmful invaders. You know from your biology classes that proteins, because they fold up into a specific three-dimensional shape, are able to very specifically recognize and bind to other organic molecules. The ligand or substrate bound by a particular protein could be a small organic molecule such as pyruvate all the way up to a large biopolymer such as a specific region of DNA, RNA, or another protein. Because they are chiral molecules, proteins are very sensitive to the stereochemistry of their ligands: a protein may bind specifically to (R)-glyceraldehyde, for example, but not bind to (S)-glyceraldehyde, just as your right hand will not fit into a left-handed baseball glove.
The over-the-counter painkiller ibuprofen is currently sold as a racemic mixture, but only the S enantiomer is effective, due to the specific way it is able to bind to and inhibit the action of prostaglandin H2 synthase, an enzyme in the body’s inflammation response process.
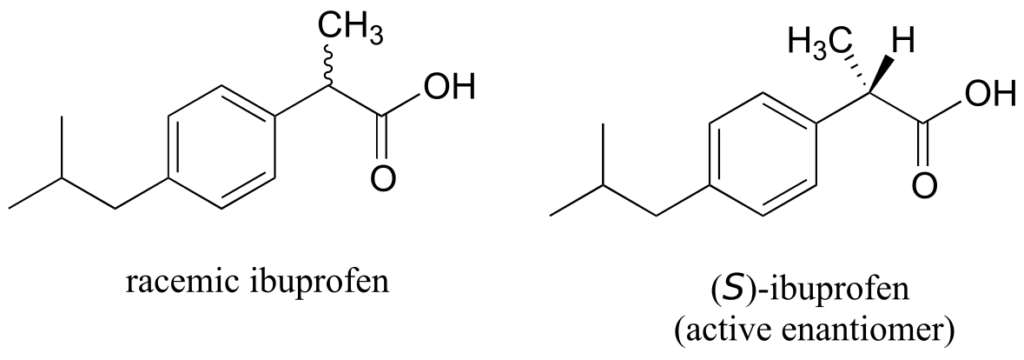
The R enantiomer of ibuprofen does not bind to prostaglandin H2 synthase in the same way as the S enantiomer, and as a consequence does not exert the same inhibitory effect on the enzyme’s action (Duggan et al., 2011, 803). Fortunately, (R)-ibuprofen apparently does not cause any harmful side effects, and is in fact isomerized gradually by an enzyme in the body to (S)-ibuprofen.
Earlier we discussed the tragic case of thalidomide, and mentioned that it appears that it is specifically the S enantiomer which caused birth defects. Many different proposals have been made over the past decades to try to explain the teratogenic (birth-defect-causing) effect of the drug, but a clear understanding still evades the scientific community. In 2010, however, a team in Japan reported evidence (“Thalidomide’s Partner in Crime,” Vogel) that thalidomide binds specifically to a protein called “thereblon.” Furthermore, when production of thereblon is blocked in female zebra fish, developmental defects occur in her offspring which are very similar to the defects caused by the administration of thalidomide, pointing to the likelihood that thalidomide binding somehow inactivates the protein, thus initiating the teratogenic effect.
You can, with a quick trip to the grocery store, directly experience the biological importance of stereoisomerism. Carvone is a chiral, plant-derived molecule that contributes to the smell of spearmint in the R form and caraway (a spice) in the S form.
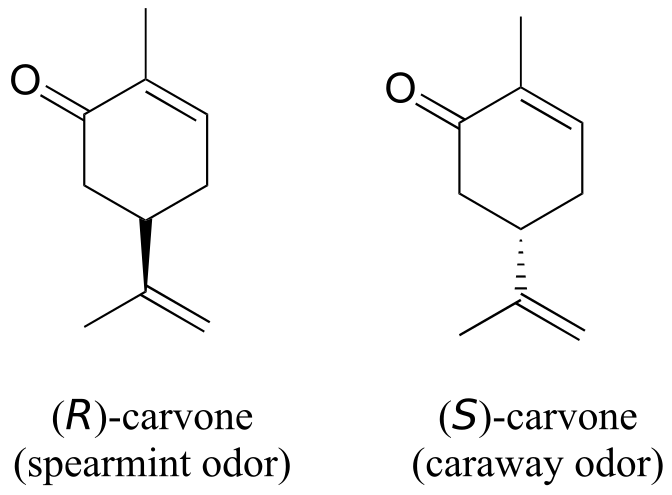
Although details are not known, the two enantiomers presumably interact differently with one or more smell receptor proteins in your nose, generating the transmission of different chemical signals to the olfactory centre of your brain.
Exercise 28:
Ephedrine, found in the Chinese traditional medicine ma huang, is a stimulant and appetite suppressant. Both pseudoephedrine and levomethamphetamine are active ingredients in over-the-counter nasal decongestants. Methamphetamine is a highly addictive and illegal stimulant, and is usually prepared in illicit “meth labs” using pseudoephedrine as a starting point.
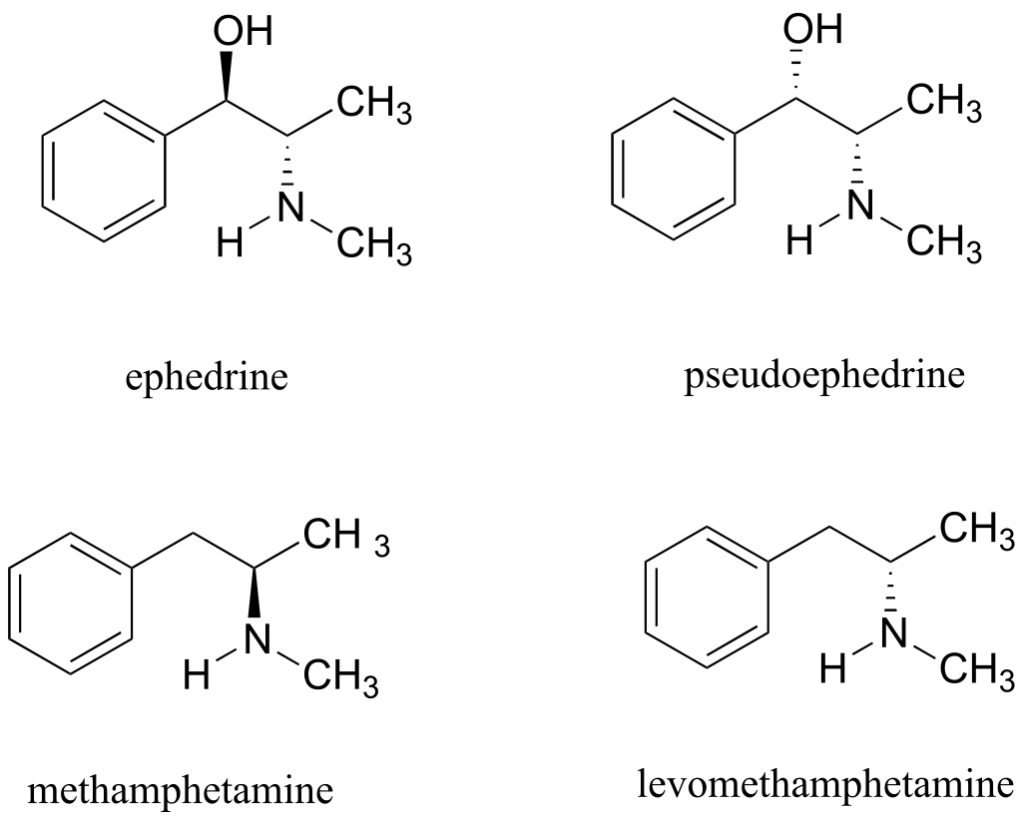
What is the relationship between ephedrine and pseudoephedrine? Between methamphetamine and levomethamphetamine? Between pseudoephedrine and methamphetamine? Your choices are: not isomers, constitutional isomers, diastereomers, enantiomers, or same molecule.
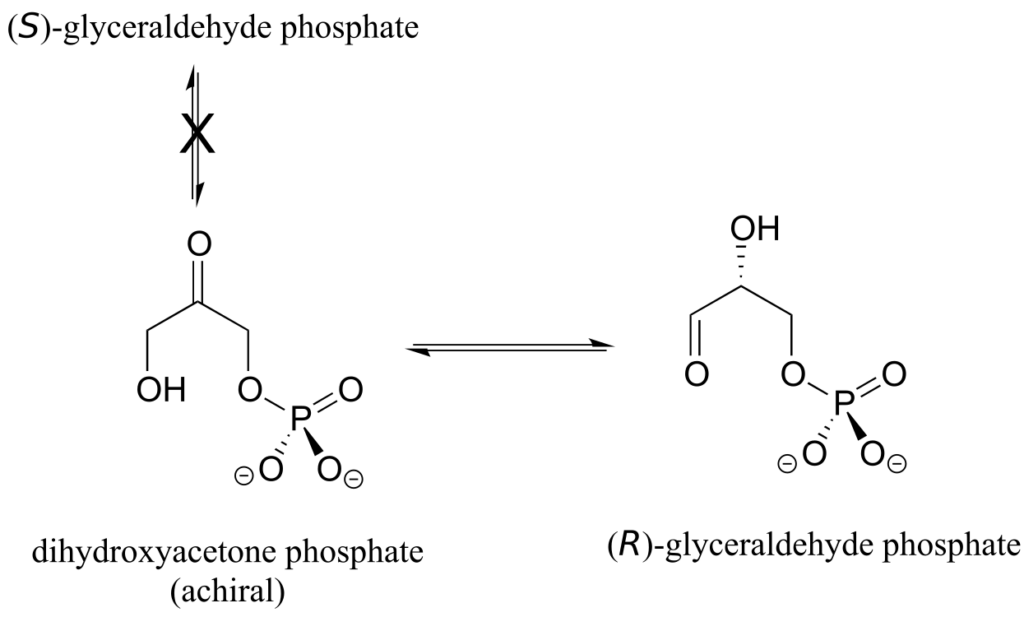
Enzymes are very specific with regard to the stereochemistry of the reactions they catalyze. When the product of a biochemical reaction contains a chiral centre or a stereogenic alkene, with very few exceptions only one stereoisomer of the product is formed. In the glycolysis pathway, for example, the enzyme triose-phosphate isomerase catalyzes the reversible interconversion between dihydroxyacetone (which is achiral) and (R)-glyceraldehyde phosphate. The (S)-glyceraldehyde enantiomer is not formed by this enzyme in the left-to-right reaction, and is not used as a starting compound in the right-to-left reaction—it does not “fit” in the active site of the enzyme.
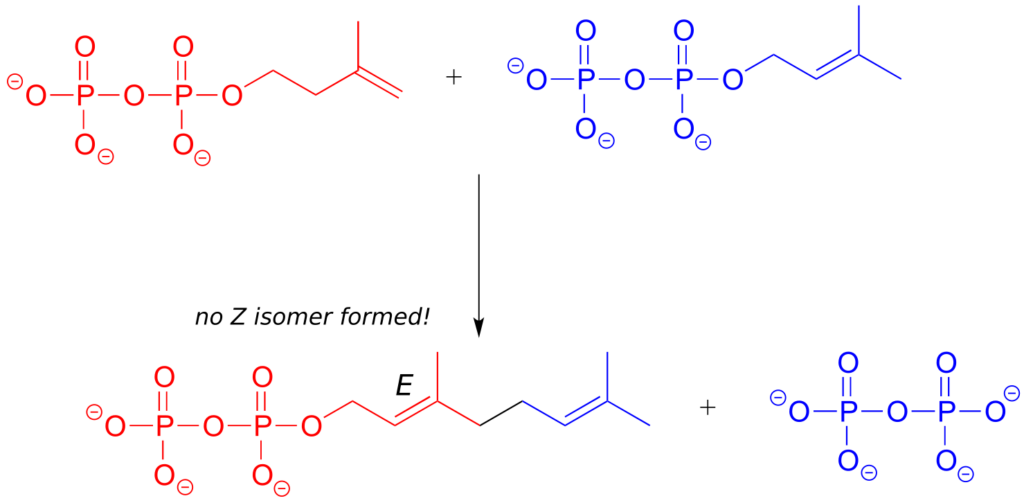
In the isoprenoid biosynthesis pathway, two five-carbon building-block molecules combine to form a ten-carbon chain containing an E-alkene group. The enzyme does not catalyze formation of the Z diastereomer.
Continuing on into your study of biological and organic chemistry, you will be learning about how enzymes are able to achieve these feats of stereochemical specificity. If you take a more advanced class in organic synthesis, you will also learn how laboratory chemists are figuring out ingenious ways to exert control over the stereochemical outcomes of nonenzymatic reactions, an area of chemistry that is particularly important in the pharmaceutical industry.
Prochirality
A: Prochiral Carbons
When a tetrahedral carbon can be converted to a chiral centre by changing only one of the attached groups, it is referred to as a “prochiral” carbon. The two hydrogens on the prochiral carbon can be described as “prochiral hydrogens.”

Note that if, in a “thought experiment,” we were to change either one of the prochiral hydrogens on a prochiral carbon centre to a deuterium (the 2H isotope of hydrogen), the carbon would now have four different substituents and thus would be a chiral centre.
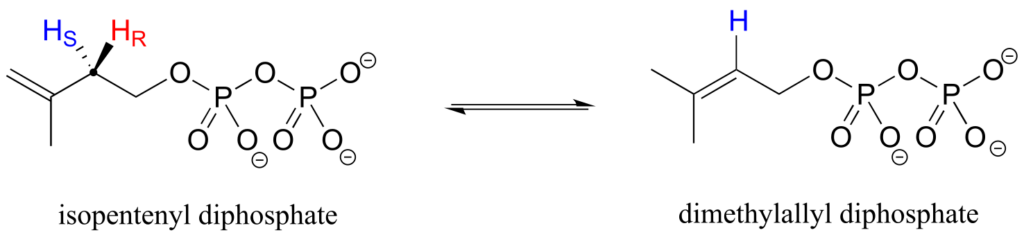
Prochirality is an important concept in biological chemistry, because enzymes can distinguish between the two “identical” groups bound to a prochiral carbon centre due to the fact that they occupy different regions in three-dimensional space. Consider the isomerization reaction below, which is part of the biosynthesis of isoprenoid compounds. We do not need to understand the reaction itself; all we need to recognize at this point is that the isomerase enzyme is able to distinguish between the prochiral “red” and the “blue” hydrogens on the isopentenyl diphosphate (IPP) substrate. In the course of the left-to-right reaction, IPP specifically loses the “red” hydrogen and keeps the “blue” one.
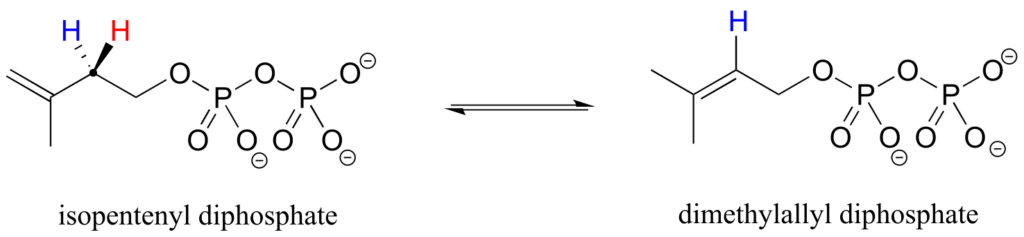
Prochiral hydrogens can be unambiguously designated using a variation on the R/S system for labelling chiral centres. For the sake of clarity, we’ll look at a very simple molecule, ethanol, to explain this system. To name the “red” and “blue” prochiral hydrogens on ethanol, we need to engage in a thought experiment. If we, in our imagination, were to arbitrarily change red H to a deuterium, the molecule would now be chiral and the chiral carbon would have the R configuration (D has a higher priority than H).

For this reason, we can refer to the red H as the pro-R hydrogen of ethanol, and label it HR. Conversely, if we change the blue H to D and leave red H as a hydrogen, the configuration of the molecule would be S, so we can refer to blue H as the pro-S hydrogen of ethanol, and label it HS.
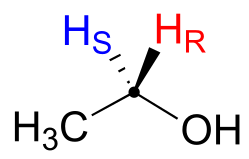
Looking back at our isoprenoid biosynthesis example, we see that it is specifically the pro-R hydrogen that the isopentenyl diphosphate substrate loses in the reaction.
Prochiral hydrogens can be designated either enantiotopic or diastereotopic. If either HR or HS on ethanol were replaced by a deuterium, the two resulting isomers would be enantiomers (because there are no other stereocentres anywhere on the molecule).
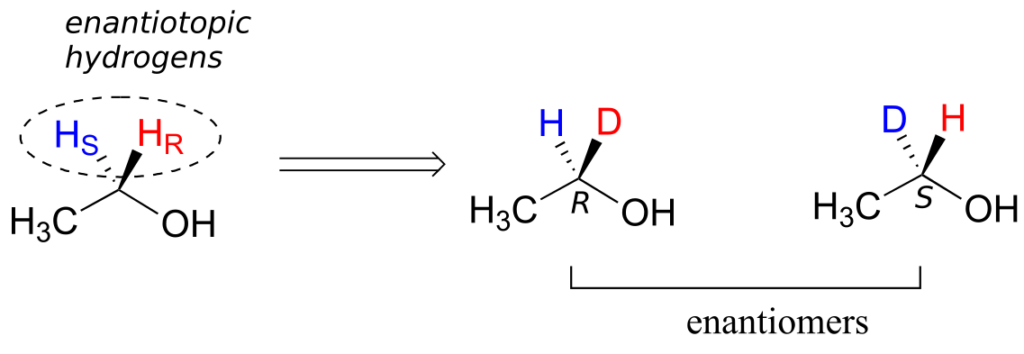
Thus, these two hydrogens are referred to as enantiotopic.
In (R)-glyceraldehyde-3-phosphate ((R)-GAP), however, we see something different:
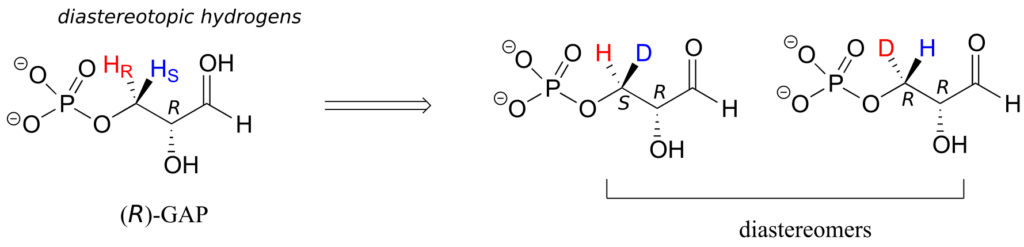
(R)-GAP already has one chiral centre. If either of the prochiral hydrogens HR or HS is replaced by a deuterium, a second chiral centre is created, and the two resulting molecules will be diastereomers (one is S,R, one is R,R). Thus, in this molecule, HR and HS are referred to as diastereotopic hydrogens.
Finally, hydrogens that can be designated neither enantiotopic nor diastereotopic are called homotopic. If a homotopic hydrogen is replaced by deuterium, a chiral centre is not created. The three hydrogen atoms on the methyl (CH3) group of ethanol (and on any methyl group) are homotopic.
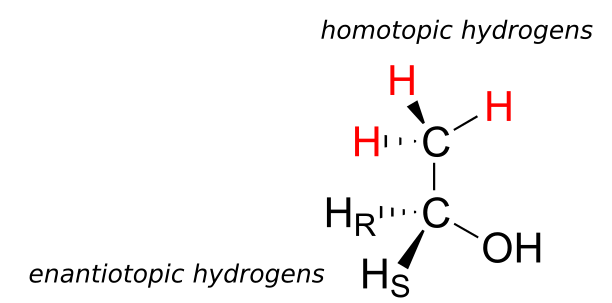
An enzyme cannot distinguish among homotopic hydrogens.
Exercise 29:
Identify in the molecules below all pairs/groups of hydrogens that are homotopic, enantiotopic, or diastereotopic. When appropriate, label prochiral hydrogens as HR or HS.
Groups other than hydrogens can be considered prochiral. The alcohol below has two prochiral methyl groups—the red one is pro-R, the blue is pro-S. How do we make these designations? Simple—just arbitrarily assign the red methyl a higher priority than the blue, and the compound now has the R configuration—therefore, red methyl is pro-R.
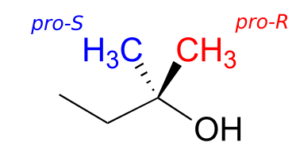
Citrate is another example. The central carbon is a prochiral centre with two “arms” that are identical except that one can be designated pro-R and the other pro-S.
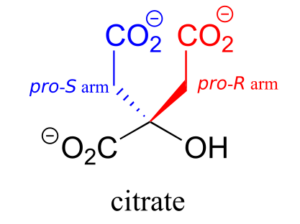
In an isomerization reaction of the citric acid (Krebs) cycle, a hydroxide is shifted specifically to the pro-R arm of citrate to form isocitrate: again, the enzyme catalyzing the reaction distinguishes between the two prochiral arms of the substrate.
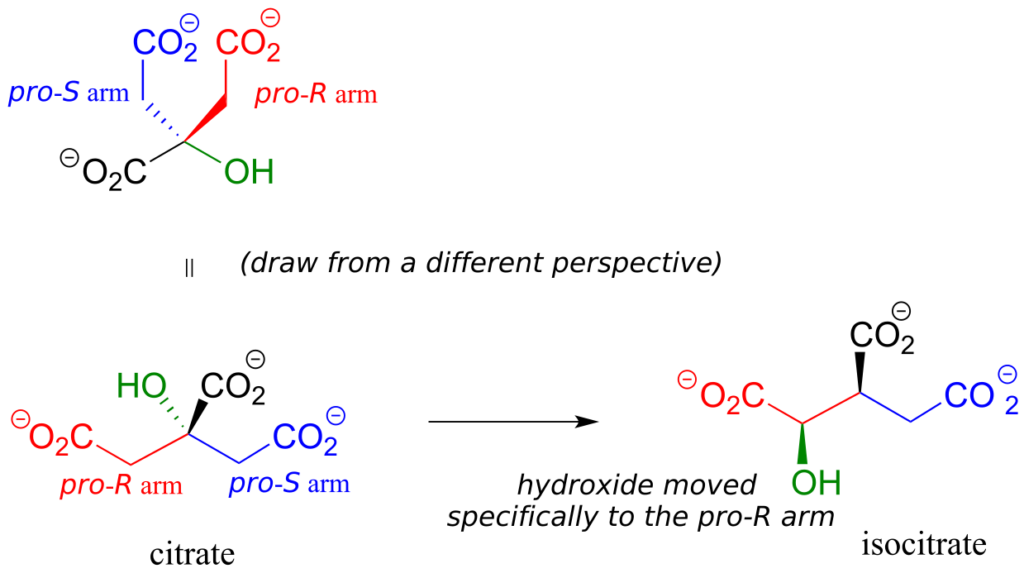
Exercise 30:
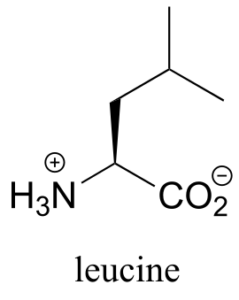
Assign pro-R and pro-S designations to all prochiral groups in the amino acid leucine. (Hint: there are two pairs of prochiral groups!). Are these prochiral groups diastereotopic or enantiotopic?
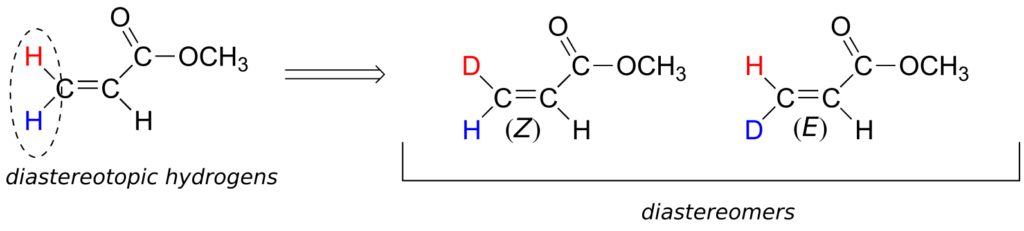
Although an alkene carbon bonded to two identical groups is not considered a prochiral centre, these two groups can be diastereotopic. Ha and Hb on the alkene below, for example, are diastereotopic: if we change one, and then the other, of these hydrogens to deuterium, the resulting compounds are E and Z diastereomers.
B: Prochiral Carbonyl and Imine Groups
Trigonal planar, sp2-hybridized carbons are not, as we well know, chiral centres—but they can be prochiral centres if they are bonded to three different substituents. We (and the enzymes that catalyze reactions for which they are substrates) can distinguish between the two planar “faces” of a prochiral sp2-hybridized group. These faces are designated by the terms re and si. To determine which is the re and which is the si face of a planar organic group, we simply use the same priority rankings that we are familiar with from the R/S system, and trace a circle: re is clockwise and si is counterclockwise.
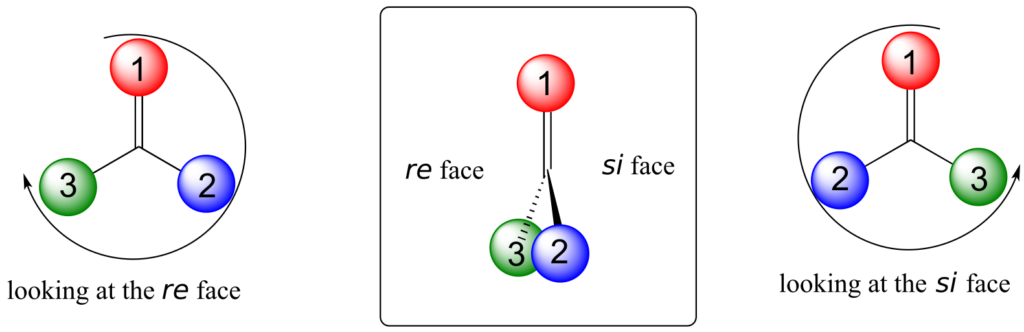
Below, for example, we are looking down on the re face of the ketone group in pyruvate:
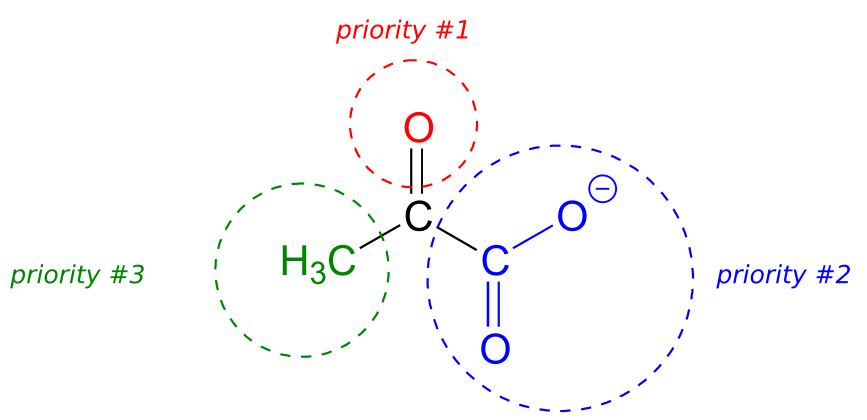
If we flipped the molecule over, we would be looking at the si face of the ketone group. Note that the carboxylate group does not have re and si faces, because two of the three substituents on that carbon are identical (when the two resonance forms of carboxylate are taken into account).
Enzymes which catalyze reactions at carbonyl carbons act specifically from one side or the other.
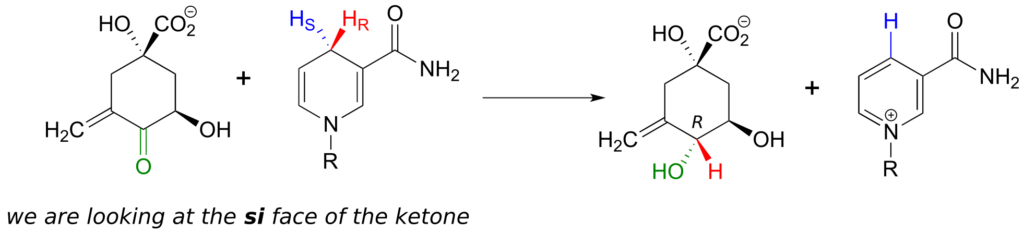
We need not worry about understanding the details of the reaction pictured above at this point, other than to notice the stereochemistry involved. The pro-R hydrogen (along with the two electrons in the C-H bond) is transferred to the si face of the ketone (in green), forming, in this particular example, an alcohol with the R configuration. If the transfer had taken place at the re face of the ketone, the result would have been an alcohol with the S configuration.
The re and si designations can also be applied to planar, sp2-hybridized carbons in alkene groups. Keep in mind that a carbon-carbon double bond has a higher priority than a carbon-carbon single bond, but a lower priority than a carbon-oxygen bond.

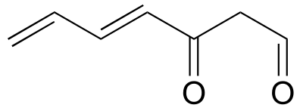
Exercise 31:
Assign a designation of re, si, or N (not prochiral) to indicate which face we are looking down on for each of the sp2-hybridized carbons in the structure below.
Summary of Key Concepts
Before you move on, you should be comfortable with the following concepts.
Conformations of Open-Chain Compounds
- Be able to distinguish between eclipsed, staggered, gauche, and anti conformations, and the rationale for trends in stability.
- Be able to draw and interpret Newman projections.
Conformations of Cyclic Compounds
- Understand the concept of angle strain in three- and four-membered rings.
- Be able to draw the envelope conformation of five-membered rings
- Be able to draw the chair and boat conformations of six-membered rings.
- In the chair conformation, be able to draw equatorial and axial substituents. Understand that large groups in the axial position experience considerable 1,3-diaxial repulsion, and thus are more stable in the equatorial position.
Stereochemistry
Hierarchy of isomeric relationships:
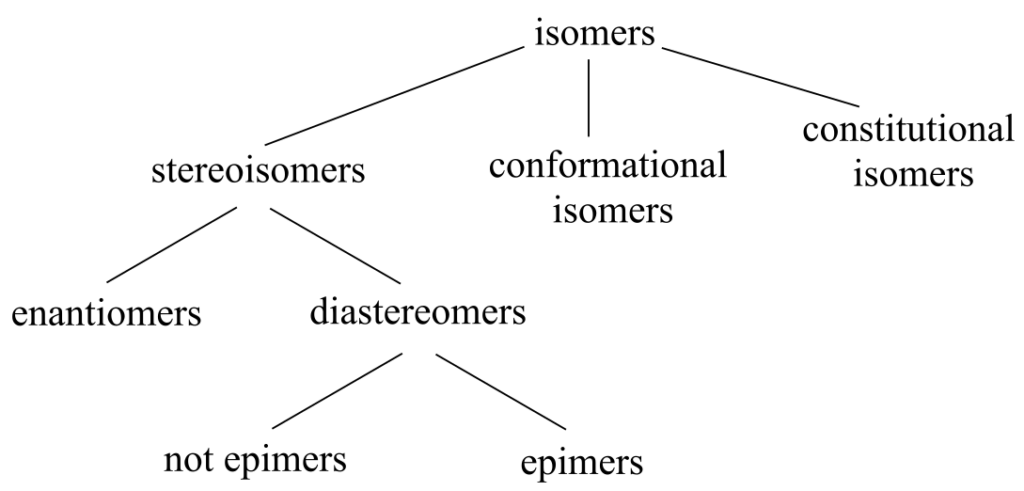
You should understand the relevant terms and concepts:
- A chiral object or molecule cannot be superimposed on its mirror image.
- A chiral centre is an sp3-hybridized (tetrahedral) carbon bonded to four different groups. A chiral centre can be labelled R or S.
- A stereogenic alkene is one in which both sides of the alkene are asymmetric, and which can therefore be labelled E or Z.
- Stereoisomers have the same molecular formula and same connectivity, but a different orientation of atoms in space.
- Enantiomers are stereoisomers which are mirror images.
- In practice, the enantiomer of a compound is the one in which all chiral centres are in the opposite configuration.
- Every chiral molecule has one and only one enantiomer.
- Achiral molecules are superimposable on their mirror image, and thus cannot have an enantiomer.
- Enantiomers have equal but opposite specific rotations, but identical physical properties otherwise.
- Diastereomers are stereoisomers which are not mirror images. They have different physical properties.
- In practice, a diastereomer of a chiral molecule with have at least one, but not all chiral centres in the opposite configuration.
- Alternatively, two diastereomers may contain a stereogenic alkene with the opposite E/Z configuration.
- A molecule has 2n-2 diastereomers, where n is the number of chiral centres plus stereogenic alkene groups. Meso compounds are an exception to this rule.
- Epimers are diastereomers which differ at only one chiral centre.
- A racemic mixture is a 50:50 mixture of two enantiomers.
- A meso compound has multiple chiral centres but, because it has a plane of symmetry, is achiral.
- You should know how to assign R/S and E/Z configuration to chiral centres and stereogenic alkenes, respectively.
- You should understand the concept of optical rotation and the definition of specific rotation.
- You should recognize that, in general, a protein can distinguish between its natural ligand and a stereoisomer of that ligand.
- You should also recognize that enzymes are highly specific with respect to stereochemistry, catalyzing the formation of only one stereoisomer of their products.
- You should be able to recognize and label pro-R and pro-S groups on prochiral tetrahetral carbons.
- You should be able to recognize re and si faces of carbonyl and imine groups.
References
Duggan, K. C., Hermanson, D. J., Musee, J., Prusakiewicz, J. J., Scheib, J. K., Carter, B. D., Banerjee, S., Oates, J. A., & Marnett, L. J. (2011). (R)-profens are substrate-selective inhibitors of endocannabinoid oxygenation by COX-2. Natural Chemical Biology, 7(11), 803–809. doi: 10.1038/nchembio.663
UMM Digital Well. (2019, July). Organic chemistry with a biological emphasis volume I. University of Minnesota. https://digitalcommons.morris.umn.edu/chem_facpubs/1/ CC BY-NC-SA 4.0.

{kind=link}
{kind=link}